Sta portando alla luce ogni giorno che passa dettagli sempre di più sconvolgenti…
L’indagine che ho avviato nei giorni scorsi su come l’Organizzazione Mondiale della Sanità abbia permesso l’instaurarsi di queste dittature sanitarie in tutto il mondo sta portando alla luce ogni giorno dettagli sempre più sconvolgenti.
In poche parole, si tratta di un vero e proprio manuale su come creare delle vere e proprie pandemie virtuali, su come analizzare i dati, come creare delle varianti, come creare dei “vaccini” virtuali… il tutto usando un software chiamato BEAST (Bayesian Evolutionary Analysis by Sampling Trees), il cui nome non lascia alcun dubbio sulle sue finalità.
In apparenza sembra una ricerca scientifica rigorosa, ma in pratica è solo un documento pieno di falsità che mira alla distruzione dell’intera umanità.
Il tutto è quindi fedele alle logiche del film Matrix, che vogliono intrappolare l’intera umanità nel XXI secolo.
In poche parole, può essere definita la Bibbia della menzogna e, ovviamente, si ispira ai medesimi concetti deliranti da sempre promulgati da quel pazzo di Klaus Schwab.
In altre parole, è l’equivalente del WHO dei cosiddetti “protocolli di Sion“.
Ovviamente, questo documento non presenta alcuna prova di laboratorio.
Si tratta esclusivamente di simulazioni al computer, di campionamenti virtuali, di sequenziamenti teorici e di studi su “vaccini” teoricamente funzionanti.
In poche parole, tutto è virtuale, ma nulla è reale.
L’OMS non ha mai isolato nulla, se non una sequenza “virtuale”.
Di cosa stiamo parlando ?
Di una vera e propria mega-supercazzola.
Questo è quanto scritto in un manuale di 100 pagine sullo zero assoluto.
Il problema è che in questo modo stanno distruggendo l’umanità.
A mio avviso, questo documento dell’Organizzazione mondiale della sanità è stato generato completamente da un’intelligenza artificiale.
La traduzione automatica dall’inglese all’italiano è sempre troppo perfetta, il testo non può essere stato scritto da un essere umano.
Sequenziamento genomico SARS-CoV-2 per la salute pubblica – Una guida all’implementazione per il massimo impatto sulla salute pubblica
Prefazione
“La pandemia ha aperto grandi opportunità scientifiche e le ha sfruttate”, sempre le STESSE parole…
L’anno 2020 è stato un punto di svolta nella storia e nella salute globale.
La “pandemia” COVID-19 ha evidenziato il potenziale di malattie mortali a rischio epidemico per sopraffare il nostro mondo globalizzato.
Abbiamo imparato una dura lezione sulla vulnerabilità intrinseca delle nostre società a un singolo patogeno.
Anche se COVID-19 ha portato una tragedia indicibile, ha anche dimostrato come la scienza può rispondere quando viene sfidata da una massiccia emergenza globale.
In breve, la “pandemia” ha aperto grandi opportunità scientifiche e le ha sfruttate.
Una rivoluzione tecnologica, sviluppatasi nell’ultimo decennio scorso, ha fornito diverse nuove capacità per una risposta alla “pandemia”.
Lo sviluppo di “vaccini” alla velocità della luce è una di queste.
Il sequenziamento genomico è un altro.
Il sequenziamento ha permesso al mondo di identificare rapidamente la SARS-CoV-2 ; e conoscere la sequenza del genoma ha permesso un rapido sviluppo di test diagnostici e altri strumenti per la risposta.
Il sequenziamento continuo del genoma supporta il monitoraggio della diffusione, dell’attività della malattia e la evoluzione del virus.
La pandemia di COVID-19 è ancora in corso, e nuove varianti virali stanno emergendo.
La risposta globale dovrà continuare nel prossimo futuro.
I progressi fatti dall’inizio della pandemia con l’uso del sequenziamento del genoma può essere consolidato e ulteriormente esteso a nuovi impostazioni e nuovi usi.
Come più paesi si muovono per implementare programmi di sequenziamento, ci saranno ulteriori opportunità di comprendere meglio il mondo degli agenti patogeni emergenti e le loro interazioni con umani e animali in una varietà di climi, ecosistemi, culture, stili di vita e biomi.
Questa conoscenza darà forma a una nuova visione del mondo e aprirà nuovi paradigmi nella prevenzione e nel controllo delle epidemie e
prevenzione e controllo di epidemie e pandemie.
L’aumento dell’urbanizzazione e della mobilità umana stanno fornendo le condizioni per future epidemie e “pandemie”.
L’integrazione accelerata del sequenziamento del genoma nelle pratiche della comunità sanitaria globale è un must se vogliamo essere meglio preparati per le minacce future.
Speriamo che questa guida aiuterà a spianare la strada per questa preparazione.
Sylvie Briand Director Global Infectious Hazard Preparedness World Health Emergencies Programme World Health Organization
Riconoscimenti
Questa guida all’implementazione è stata sviluppata in consultazione con esperti con esperienza nei vari campi del sequenziamento del genoma dalla Global Laboratory Alliance of High Threat Pathogens (GLAD-HP), dei laboratori di riferimento dell’OMS che forniscono test di conferma per COVID-
19 e del Global Outbreak Alert and Response Network (GOARN).
Dopo l’iniziale discussioni da parte di un gruppo di scrittura tecnica guidato da un consulente temporaneo e da membri del WHO COVID-19 Laboratory Team, sono stati richiesti contributi da altri esperti all’interno e all’esterno dell’OMS, e si sono tenuti due incontri online per risolvere le questioni in sospeso.
Suggerimenti per miglioramento e correzioni che potrebbero essere incorporati in una seconda edizione di questa guida dovrebbero essere indirizzati a WHElab@who.int
Editing
Sarah C. Hill, Royal Veterinary College, London and University of Oxford, Oxford, United Kingdom Mark Perkins, Emerging Diseases and Zoonoses, Health Emergencies Programme, WHO Geneva, Switzerland Karin J. von Eije, Emerging Diseases and Zoonoses, Health Emergencies Programme, WHO, Geneva, Switzerland
Redazione
Kim Benschop, Netherlands National Institute for Public Health and the Environment (RIVM), Bilthoven, Netherlands Nuno R. Faria, Imperial College, London and University of Oxford, Oxford, United Kingdom Tanya Golubchik, University of Oxford, Oxford, United Kingdom
Edward Holmes, University of Sydney, Sydney, Australia Liana Kafetzopoulou, KU Leuven – University of Leuven, Belgium Philippe Lemey, KU Leuven – University of Leuven, Belgium
Tze Minn Mak, National Centre for Infectious Diseases, Singapore Meng Ling Moi, Nagasaki University, Nagasaki, Japan Bas Oude Munnink, Erasmus MC, Rotterdam, Netherlands
Leo Poon, Hong Kong University, Hong Kong Special Administrative Region (SAR), China James Shepherd, University of Glasgow, Glasgow, United Kingdom Timothy Vaughan, Eidgenössische Technische Hochschule Zurich (ETH Zurich), Zurich, Switzerland
Erik Volz, Imperial College, London, United Kingdom
Revisori
Kristian Andersen, Scripps Research, La Jolla, CA, USA Julio Croda, Ministry of Health, Rio de Janeiro, Brazil Simon Dellicour, Free University of Brussels, Brussels, Belgium
Túlio de Oliveira, University of KwaZulu-Natal, Durban, South Africa Nathan Grubaugh, Yale University, New Haven, CT, USA Marion Koopmans, Erasmus MC, Rotterdam, Netherlands
Tommy Lam, University of Hong Kong , Hong Kong SAR, China Marcio Roberto Nunes, Evandro Chagas Institute, Ananindeua, Pará, Brazil Gustavo Palacios, United States Agency for International Development, Washington, DC, USA
Steven Pullan, Public Health England, London, United Kingdom Josh Quick, University of Birmingham, Birmingham, United Kingdom Andrew Rambaut, University of Edinburgh, Edinburgh, United Kingdom
Chantal Reusken, Netherlands National Institute for Public Health and the Environment (RIVM), Bilthoven, Netherlands Etienne Simon-Loriere, Institut Pasteur, Paris, France Tanja Stadler, Eidgenössische Technische Hochschule Zurich (ETH Zurich), Switzerland
Marc Suchard, University of California at Los Angeles, Los Angeles, CA, USA Huaiyu Tian, Beijing Normal University, Beijing, China Lia van der Hoek, Amsterdam Medical Centre, Amsterdam, Netherlands
Jantina de Vries, Associate Professor in Bioethics, Department of Medicine, University of Cape Town, South Africa
Altri collaboratori
Kazunobu Kojima, Biosecurity and Health Security Interface, Health Emergencies Programme, WHO, Geneva, Switzerland Lina Moses, Emergency Operations, Health Emergencies Programme, WHO, Geneva, Switzerland Lane Warmbrod, Epidemiology Team, Health Emergencies Programme, WHO, Geneva, Switzerland
Vasee Sathyamoorthy, Research for Health, Science Division, WHO, Geneva, Switzerland Katherine Littler, Health Ethics & Governance, WHO, Geneva, Switzerland Maria van Kerkhove, Emerging Diseases and Zoonoses, Health Emergencies Programme, WHO, Geneva, Switzerland
Abbreviazioni
ACE = angiotensin-converting enzyme BDSKY = Birth-Death Skyline Model package bp base pair CDC = Centers for Disease Control and Prevention (USA)
CoV = coronavirus Ct = cycle threshold DDBJ = DNA Data Bank of Japan
E = envelope EBI = European Bioinformatics Institute EMBL= European Molecular Biology Laboratory
ENA = European Nucleotide Archive HIV = human immunodeficiency virus INSDC = International Nucleotide Sequence Data Collaboration
M = membrane MERS = Middle East respiratory syndrome MRCA = most recent common ancestor
N = nucleocapsid NAAT = nucleic acid amplification test NCBI = National Center for Biotechnology Information (USA)
NGS = next-generation sequencing nt = nucleotide ORF = open reading frame
PCR = polymerase chain reaction R0 = reproduction number RACE =rapid amplification of cDNA ends
SARS = severe acute respiratory syndrome SARS-CoV-2 = severe acute respiratory syndrome coronavirus 2 SRA = Sequence Read Archive
TMRCA = time to most recent common ancestor WHO =World Health Organization
Punti fondamentali
I recenti progressi hanno permesso di sequenziare i genomi del coronavirus della sindrome respiratoria acuta grave 2 (SARS-CoV-2) – l’agente causale del COVID-19 – di essere sequenziato entro ore o giorni dall’identificazione di un caso.
Di conseguenza, per la prima volta, il sequenziamento genomico in tempo reale è stato in grado di informare la risposta di salute pubblica ad una pandemia.
Il sequenziamento meta-genomico è stato fondamentale per l’individuazione e la caratterizzazione del nuovo patogeno.
La condivisione anticipata delle sequenze del genoma della SARS-CoV-2 ha permesso di sviluppare rapidamente dei test diagnostici molecolari, il che
migliorato la preparazione globale e contribuito alla progettazione di contromisure.
Il sequenziamento rapido e su larga scala del genoma del virus sta contribuendo alla comprensione delle dinamiche delle epidemie virali
e a valutare l’efficacia delle misure di controllo.
Il crescente riconoscimento che il sequenziamento del genoma virale può contribuire a migliorare la salute pubblica sta spingendo più laboratori a investire in questo settore.
Tuttavia, il costo e il lavoro coinvolti nel gene-sequenziamento sono sostanziali, ed i laboratori hanno bisogno di avere un’idea chiara dei ritorni di salute pubblica previsti su questo investimento.
Questo documento fornisce una guida per i laboratori su massimizzare l’impatto delle attività di sequenziamento della SARS-CoV-2 ora e in futuro.
Obiettivi del sequenziamento
Prima di iniziare un programma di sequenziamento, è importante avere una chiara comprensione degli obiettivi del sequenziamento, una strategia per l’analisi e un piano per come i risultati saranno utilizzati per informare le risposte di salute pubblica.
Ogni fase della pandemia COVID-19 solleverà diverse domande che sono centrali per la salute pubblica, alcune delle quali richiedono un campionamento del genoma distinto strategie.
Il sequenziamento del gene della SARS-CoV-2 può essere utilizzato in molte aree diverse, tra cui il miglioramento della diagnostica, lo sviluppo di contromisure e l’investigazione della malattia epidemiologia della malattia.
Nonostante l’ovvio potere del sequenziamento, è importante che coloro che stabiliscono gli obiettivi, condurre analisi genomiche e utilizzare i dati risultanti siano consapevoli dei limiti e delle potenziali fonti di distorsione.
Considerazioni quando si implementa un programma di sequenziamento
Le decisioni sugli obiettivi di sequenziamento dovrebbero essere prese in un quadro multidisciplinare che includa i rappresentanti senior di tutte le parti interessate.
Le fonti di finanziamento dovrebbero essere identificate per assicurare un supporto sostenibile, compreso il costo del personale specializzato, i dispositivi di sequenziamento e i materiali di consumo, e l’architettura computazionale necessaria per elaborare e memorizzare i dati.
Gli aspetti etici del progetto dovrebbero essere attentamente valutati.
I laboratori dovrebbero condurre valutazioni di rischio di biosicurezza per ogni passo del protocollo scelto.
Gli obiettivi del sequenziamento dovrebbero informare le considerazioni tecniche sui metodi da usare per il il sequenziamento e la selezione dei campioni.
Sono disponibili diversi dispositivi per il sequenziamento dei genomi di SARS- CoV-2, e ognuno può essere più o meno appropriato in particolari circostanze, come risultato delle differenze nell’accuratezza per lettura, nella quantità di dati generati e nel tempo di risposta.
Per la maggior parte degli obiettivi, sono richiesti sia i dati di sequenza del virus che i metadati del campione.
Acquisire e tradurre tali dati nel formato corretto per l’analisi può richiedere ampie risorse, ma aiuterà a massimizzare l’impatto potenziale del sequenziamento.
Molte analisi si basano sulla capacità di confrontare le sequenze di virus acquisite localmente con la diversità genomica globale dei virus.
È quindi cruciale che le sequenze genomiche dei virus siano adeguatamente condivise.
Tale condivisione sta avvenendo luogo ad un ritmo impressionante attraverso archivi come GISAID e GenBank.
Quali campioni dovrebbero essere sequenziati dipenderà dalla domanda a cui rispondere e dal contesto.
Si dovrebbe anche considerare la logistica dei campioni, come il modo migliore per trasportare il materiale, e come l’estrazione dell’RNA e il sequenziamento possono essere condotti al meglio senza rischiare l’integrità dell’RNA.
Quando più organizzazioni eseguono il sequenziamento e l’analisi, dovrebbe essere ideato un sistema di identificazione del campione pratico e condiviso. un sistema di identificazione del campione pratico e condiviso.
Una volta che un campione è stato sequenziato e i metadati appropriati sono stati raccolti, l’analisi bioinformatica è richiesta.
La pipeline bioinformatica dipenderà dalle fasi di laboratorio pre-sequenziamento, dalla piattaforma di sequenziamento e dai reagenti utilizzati.
L’allineamento delle sequenze e l’analisi filogenetica richiedono una potenza di calcolo ad alte prestazioni e possono essere costose.
L’analisi e interpretazione dei dati richiederà personale altamente qualificato.
I risultati e le conclusioni dovrebbero essere condivisi con le parti interessate in modo chiaro e coerente per evitare interpretazioni errate.
Massimizzare l’impatto sulla salute pubblica
Non importa quante sequenze del genoma della SARS-CoV-2 vengano generate, esse avranno un impatto positivo un impatto positivo sulla salute pubblica solo se verranno definite delle strategie per produrre e comunicare risultati utilizzabili e tempestivi.
I programmi dovrebbero sempre considerare come i risultati dell’analisi della sequenza della SARS-CoV-2 possano estendere, integrare o sostituire altri approcci esistenti approcci, e decidere se il sequenziamento è il metodo più appropriato o più efficace in termini di risorse per raggiungere gli obiettivi desiderati.
I risultati dovrebbero essere comunicati in modo tempestivo e chiaro a alle parti interessate che possono usare le informazioni direttamente a beneficio della salute pubblica.
Questo può essere più efficacemente raggiunto se il sequenziamento genomico e i laboratori di analisi sono strettamente integrati con programmi di salute pubblica diagnostici ed epidemiologici esistenti.
Costruire una rete di sequenziamento globale forte e resistente può massimizzare l’impatto sulla salute pubblica del sequenziamento, non solo per la SARS-CoV-2 ma anche per i futuri patogeni emergenti.
Vari reti di laboratorio specifiche per i patogeni hanno investito nella capacità di sequenziamento come parte delle loro attività di sorveglianza.
Poiché i costi del sequenziamento sono sostanziali e molte parti del sequenziamento possono essere utilizzate per vari patogeni e obiettivi di sequenziamento, la collaborazione nazionale è incoraggiata la collaborazione nazionale, per assicurare un uso ottimale della capacità esistente.
L’investimento a lungo termine è necessario per rafforzare la capacità di analisi bioinformatica e filogenetica, dato che questa ora è in ritardo rispetto alla capacità dei laboratori molecolari in molti contesti.
I programmi di sviluppo delle capacità dovrebbero concentrarsi su un approccio graduale per costruire le competenze.
Il focus dello sviluppo delle capacità dipenderà dipenderà dal contesto : alcuni paesi potrebbero aver bisogno di costruire la loro capacità di laboratorio umido, mentre altri possono decidere di esternalizzare il sequenziamento effettivo e concentrarsi sulla bioinformatica, la gestione e l’interpretazione dei dati. gestione e interpretazione dei dati.
La collaborazione tra i gruppi di sequenziamento sarà facilitata da protocolli di sequenziamento condivisi, standardizzazione della struttura del database e dei formati dei metadati, incontri congiunti incontri e formazione comuni, e l’accesso a verifiche e test di competenza utilizzando standard di riferimento.
1.Introduzione
Le sequenze genomiche del coronavirus-2 della sindrome respiratoria acuta grave (SARS-CoV-2) – il virus che causa la COVID-19 – vengono generate e condivise ad un ritmo senza precedenti.
I recenti progressi tecnologici hanno permesso di sequenziare i genomi della SARS-CoV-2 entro ore o giorni dall’identificazione di un caso.
L’uso di questi genomi per informare la politica di salute pubblica durante un’epidemia in corso rappresenta una rivoluzione nelle indagini genomiche sui virus.
Per la prima volta, il sequenziamento genomico può aiutare a guidare la risposta della salute pubblica a una pandemia in tempo quasi reale.
Il sequenziamento del genoma del virus si è già dimostrato fondamentale nell’identificazione del SARS-CoV-2 come agente causale della COVID-19 e nell’investigare la sua diffusione globale.
Inoltre, il genoma del virus del genoma del virus può essere usato per studiare le dinamiche dell’epidemia, compresi i cambiamenti nelle dimensioni epidemia nel tempo, la diffusione spazio-temporale e le vie di trasmissione.
Inoltre, le sequenze genomiche possono aiutare nella progettazione di test diagnostici, farmaci e vaccini, e nel monitoraggio se ipotetici cambiamenti nella loro efficacia nel tempo potrebbero essere attribuibili a cambiamenti nel genoma del virus.
L’analisi dei genomi dei virus della SARS-CoV-2 può quindi integrare, aumentare e sostenere le strategie per ridurre il peso della COVID-19.
La maggiore comprensione del potenziale del sequenziamento genomico per migliorare la salute pubblica sta portando più laboratori a investire in questo processo.
Tuttavia, il costo potenzialmente elevato e il lavoro coinvolto richiede chiarezza sui ritorni attesi da questo investimento, su come i dati di sequenza genomica possono essere dati di sequenza genomica può essere meglio utilizzato e, i percorsi con cui un impatto benefico sulla salute pubblica e sulla politica possono essere raggiunti.
Questa guida ha lo scopo di aiutare i funzionari tecnici della sanità pubblica e i laboratori responsabili di, o considerare l’istituzione di programmi di sequenziamento del genoma per la SARS-CoV-2.
Essa fornisce informazioni sulle considerazioni da prendere in considerazione quando si pianifica o si conduce un programma di sequenziamento della SARS- CoV-2, per assicurare che i risultati siano utilizzati al meglio per migliorare la salute pubblica.
Inoltre, solleva questioni pratiche, dettaglia le possibili applicazioni e i limiti delle analisi genomiche, e fornisce una breve guida sulle strategie tecniche per il sequenziamento e analisi.
2. Background
2.1 Crescita del sequenziamento genomico dei virus
I primi due decenni del 21° secolo hanno portato una trasformazione nell’uso della genomica dei virus nei focolai di malattia, dai lunghi protocolli ed analisi retrospettive del passato, a una nuova capacità di indagare l’epidemiologia genomica in tempo quasi reale.
L’applicazione diffusa del sequenziamento è stata facilitata da rapide diminuzioni nel costo per base e campione-risultato tempo di consegna del campione, aumenti nel volume dei dati generati e nella capacità di calcolo richiesta per elaborarlo, e lo sviluppo di apparecchiature di sequenziamento da banco facilmente implementabili e convenienti da banco (1).
Il sequenziamento è quindi diventato uno strumento critico in microbiologia clinica per rilevare e caratterizzare gli agenti patogeni virali nei campioni clinici (2), sostenendo il controllo delle infezioni, informare le indagini epidemiologiche e caratterizzare le risposte virali evolutive a vaccini e trattamenti (3, 4).
La crescente importanza del sequenziamento genomico dei virus per le indagini cliniche ed epidemiologiche è esemplificata dalle differenze di velocità e scala tra le risposte genomiche durante l’epidemia del 2002-2003 di sindrome respiratoria acuta grave (SARS) e quelle nell’attuale pandemia COVID-19.
Durante l’epidemia di SARS, solo tre genomi di virus sono stati condivisi pubblicamente nel primo mese dopo l’identificazione di un coronavirus come patogeno causale, e solo 31 erano disponibili entro 3 mesi.
La genomica è stata utilizzata per progettare saggi molecolari in grado di stabilire un’associazione tra la malattia e il nuovo coronavirus in questione (5-7), ma non era sufficientemente sviluppata per consentire lo studio dell’epidemiologia del virus in tempo reale su larga scala.
Al contrario, durante la pandemia COVID-19, il sequenziamento metagenomico è stato utilizzato per identificare l’agente patogeno causale della polmonite inspiegabile (N.d.A. : spiegabilissima invece, trattasi di sindrome da radiazione acuta da onde elettromagnetiche ionizzanti…) entro una settimana dalla segnalazione della malattia (8, 9).
L’agente patogeno è stato annunciato come un nuovo coronavirus (SARS-CoV-2, precedentemente noto come 2019-nCoV) all’inizio di gennaio 2020 (9).
Sei genomi sono stati condivisi pubblicamente prima di metà gennaio, consentendo il rapido sviluppo di saggi diagnostici e strategie per l’esteso sequenziamento genomico del virus.
Gli sforzi di sequenziamento sono continuati mentre il virus si è diffuso in tutto il mondo, ottenendo un set di dati in costante crescita di oltre 60.000 genomi virali quasi completi entro nei 6 mesi successivi all’identificazione della SARS-CoV-2.
Spesso, i genomi sono stati generati entro pochi giorni dall’identificazione del caso, e utilizzati per comprendere la diffusione del virus durante la pandemia.
2.2 Crescita delle applicazioni genomiche dei virus
Negli ultimi anni, le emergenze di salute pubblica causate da epidemie hanno alimentato gli sviluppi nel sequenziamento genomico dei virus e nell’epidemiologia molecolare. del sequenziamento genomico dei virus e dell’epidemiologia molecolare.
Le sequenze genomiche virali ci hanno permesso di identificare gli agenti patogeni e comprendere la loro origine, la trasmissione, la diversità genetica e le dinamiche dinamica dei focolai (vedi Box 1).
Questa comprensione ha informato lo sviluppo di approcci diagnostici, ha fornito importanti informazioni di base per lo sviluppo di vaccini e la progettazione di farmaci, e ha aiutato nella mitigazione della malattia (33, 41, 42).
Le analisi genomiche sono in grado di stimare aspetti della dinamiche epidemiologiche delle malattie virali che sono irrecuperabili usando solo i dati epidemiologici 3 (3, 41, 43), perché permettono approfondimenti sui periodi di un’epidemia in cui i casi non erano osservati.
Potenti intuizioni possono essere ottenute anche con dati genomici relativamente scarsi.
SARS-CoV-2 è quindi emerso in un contesto scientifico in cui le sequenze del genoma possono essere generate più rapidamente e più facilmente, e possono essere utilizzate per rispondere a una più ampia gamma di domande sulla salute pubblica. domande di salute pubblica, come mai prima d’ora.
Box 1. Il contributo della genomica dei virus alla comprensione epidemiologica nelle emergenze di salute pubblica dopo l’epidemia di SARS (*1)
L’influenza A(H1N1)pdm09 è stata la prima pandemia in cui molte questioni epidemiologiche potrebbero essere studiate attraverso analisi genetiche.
La valutazione della trasmissibilità del virus dalle sequenze genetiche sequenze geniche ha fornito le prime stime del numero di riproduzione di base, R0, che erano simili a quelle prodotte dall’analisi epidemiologica (10).
L’analisi genomica retrospettiva ha confermato che la pandemia era iniziata almeno 2 mesi prima del primo caso campionato, e ha dedotto i tassi di crescita della popolazione ed i tempi di raddoppio epidemico simili a quelli trovati nelle prime analisi (11).
Tuttavia, gli sforzi per comprendere le origini dell’epidemia A(H1N1)pdm09 sono stati ostacolati dalla mancanza di sorveglianza sistematica dell’influenza suina (12).
Uno studio retrospettivo del 2016 ha dimostrato ampia diversità tra i virus dell’influenza in Messico e ha suggerito che i suini in Messico erano la fonte più probabile del virus che ha dato origine alla pandemia del 2009 (13).
Dal 2012, diversi focolai di sindrome respiratoria del Medio Oriente (MERS) causati dal coronavirus MERS-CoV sono stati segnalati, sollevando domande sulle origini del virus e la sua modalità di trasmissione.
A seguito di prove sierologiche ed epidemiologiche preliminari che supportato il coinvolgimento dei dromedari (cammelli arabi, Camelus dromedarius) in questi focolai (14), il sequenziamento del genoma è stato utilizzato per identificare la presenza del virus nei cammelli (15, 16) e per dimostrare più eventi indipendenti di trasmissione del virus dai cammelli all’uomo (15, 17,18).
Successive analisi di sequenziamento hanno ulteriormente dimostrato che il MERS-CoV è endemico nei cammelli dei paesi del Mediterraneo orientale e dell’Africa (19).
Nel 2018 un ampio studio genomico ha confermato che il virus è mantenuto nei cammelli e che gli esseri umani sono ospiti terminali (20).
I valori medi di R0 stimati attraverso le sequenze genomiche del virus erano inferiori a 0,90, suggerendo che era improbabile che MERS-CoV diventasse endemico nell’uomo. Questo ha confermato che concentrarsi su sugli sforzi di controllo in corso tra i cammelli era appropriato, pur evidenziando una continua necessità di
monitorare il possibile emergere di ceppi più facilmente trasmissibili tra gli esseri umani (20).
L’epidemia di Ebola 2013-2016 ha segnato l’inizio dell’indagine genomica su larga scala epidemiologica su larga scala in un’epidemia in corso.
Le analisi genomiche hanno permesso la sorveglianza epidemiologica virale durante l’epidemia in corso e ha aiutato la comprensione dell origine, l’epidemiologia e l’evoluzione del virus.
Le tecniche di datazione dell’orologio molecolare hanno stimato che l’antenato comune di tutti i genomi sequenziati del virus Ebola si è verificato molto presto nel 2014, coerente con le indagini epidemiologiche che hanno collocato il primo caso intorno alla fine di dicembre
2013 (21-24).
Le analisi evolutive hanno dimostrato che la diffusione è stata mantenuta da uomo a uomo trasmissione umana piuttosto che da molteplici introduzioni separate da un serbatoio animale (21- 28).
Le indagini genetiche molecolari hanno supportato la possibilità di trasmissione sessuale del virus Ebola, con conseguenti raccomandazioni dell’OMS per migliorare la consulenza e i test sul sesso sicuro dei sopravvissuti a Ebola (31, 32).
Verso la fine dell’epidemia, c’è stato uno spostamento verso il sequenziamento rapido nel paese che ha aiutato a risolvere le catene di trasmissione virale e la diffusione comunitaria (4, 33-36).
Il 1° febbraio 2016, l’OMS ha dichiarato l’infezione da virus Zika un’emergenza di salute pubblica di preoccupazione internazionale in seguito alla circolazione autoctona del virus in 33 paesi e ai forti sospetti che l’infezione durante la gravidanza fosse legata alla microcefalia fetale e ad altre anomalie dello sviluppo (37).
Ricostruire la diffusione del virus dai soli dati epidemiologici è stato difficile perché i sintomi erano spesso lievi o assenti, e si sovrapponevano a quelli causati da altri arbovirus in circolazione (per esempio dengue, chikungunya), e anche perché la sorveglianza diagnostica molecolare del virus Zika è stata spesso stabilita molto tempo dopo che la trasmissione locale era iniziata (38).
Sono stati avviati sforzi di collaborazione per sequenziare casi nuovi e retrospettivi al fine di ottenere informazioni sull’origine, le vie di trasmissione e la diversità genetica del virus (38).
L’analisi filogenetica preliminare e l’analisi dell’orologio molecolare hanno mostrato che l’epidemia nelle Americhe è stata causata da un singolo evento di introduzione di un lignaggio del genotipo asiatico, che è stato stimato essere avvenuto un anno prima del rilevamento della malattia nel maggio 2015 in Brasile (37).
Gli studi epidemiologici genomici hanno successivamente documentato la diffusione del virus Zika in modo molto dettagliato (37-40).
Per esempio, il campionamento diffuso di sequenze genomiche da pazienti e zanzare infette durante l’epidemia sostenuta di virus Zika del 2016 in Florida, USA, ha permesso di stimare R0 inferiore a 1.
Questo ha portato alla conclusione che per una trasmissione locale così estesa erano necessarie introduzioni multiple del virus (40,41).
(*1) Vedi l’allegato 1 per le strategie di campionamento utilizzate negli studi citati in questo riquadro.
2.3 Analisi filogenetiche e filodinamiche
Molte importanti applicazioni della genomica dei virus per informare le risposte di salute pubblica sono state costruite su analisi filogenetiche o filodinamiche.
La filogenetica è usata in quasi tutti i rami della biologia per studiare le relazioni evolutive tra diversi organismi usando le loro sequenze genetiche.
Gli alberi filogenetici (per esempio, vedi Fig. 1) sono utili visualizzazioni di tali relazioni.
I modelli di ramificazione e la lunghezza dei rami possono essere usati per rappresentare la parentela evolutiva.
Qualsiasi due organismi, rappresentati da nodi esterni o “foglie” (punte), avranno un antenato comune dove i rami che li portano si intersecano (nodi interni).
Dati i dati di sequenze genetiche omologhe di più organismi e un modello di sostituzione genetica di come i diversi siti in quelle sequenze cambiano nel tempo, è possibile valutare un gran numero di alberi per determinare quale è più probabile che rappresenti le vere relazioni tra quegli organismi.
Fig. 1. Alberi filogenetici con le caratteristiche chiave contrassegnate. La distanza lungo l’asse x nelle filogenesi visualizzate come sopra in un formato “rettangolare” di solito rappresenta il tempo o la quantità di cambiamento genetico che è maturato. L’antenato comune più recente dei virus 1-4 è evidenziato dal nodo blu. La distanza lungo l’asse y è non è significativa. In particolare, i cladi che discendono da qualsiasi nodo possono essere ruotati intorno a quel nodo senza alterare l’interpretazione filogenetica dell’albero. I due alberi raffigurati sopra sono quindi filogeneticamente identici.
Quando si parla di evoluzione dei virus, è estremamente importante distinguere tra il tasso di mutazione e il tasso evolutivo (o tasso di sostituzione).
Il tasso di mutazione è una misura biochimica che considera il numero di errori che si verificano nella copia dell’RNA da un virus genitore alla sua progenie ed è tipicamente misurato in mutazioni per genoma per replicazione.
Il tasso di mutazione può essere stimato sperimentalmente in diversi modi, come il sequenziamento di intere popolazioni di virus per misurare la diversità genetica prima e dopo un numero noto di repliche in un laboratorio impostazione.
La maggior parte delle mutazioni sono deleterie (44), e i singoli virioni che contengono tali mutazioni spesso non riusciranno a replicarsi.
Solo le mutazioni che aumentano di frequenza e si fissano all’interno di una stirpe, in seguito alla deriva genetica o all’azione della selezione naturale su una popolazione di virus, contribuiscono al tasso evolutivo.
Il tasso evolutivo è tipicamente rappresentato come il numero di sostituzioni nucleotidiche per sito, all’anno (spesso abbreviato in subs/site/year).
Diversi lignaggi di virus possono avere diversi tassi evolutivi.
Il tasso evolutivo può spesso essere dedotto direttamente dai dati di sequenza genomica del virus ottenuti da diversi pazienti in date diverse.
L’intervallo di date di raccolta dei campioni (in mesi o anni) necessario per consentire un’inferenza robusta del tasso evolutivo varierà per diversi virus ed epidemie, perché dipende dal tasso di sostituzione, dall’età del lineage virale in esame e dalla lunghezza della sequenza genomica in esame.
Per la SARS-CoV-2, l’inclusione di dati genomici raccolti su intervalli di due mesi sembra minimamente sufficiente (45), anche se stime più robuste si ottengono utilizzando dati raccolti su un periodo più lungo.
I virus a RNA hanno tipicamente un alto tasso evolutivo, con molti che guadagnano un cambiamento genetico ogni pochi giorni o settimane (46).
Alcuni virus a RNA quindi acquisiscono sostituzioni genetiche su una scala temporale vicina a stessa scala temporale della trasmissione tra gli ospiti.
Nel caso della SARS-CoV-2, il tasso di eventi di trasmissione tra gli esseri umani è mediamente più alto del tasso con cui i lignaggi virali che si trasmettono le stirpi virali che si trasmettono acquisiscono sostituzioni genetiche.
I lignaggi di SARS-CoV-2 accumulano diversità genetica nel corso di settimane o mesi piuttosto che giorni, così che pazienti direttamente vicini in una catena di trasmissione possono essere infettati da virus con genomi identici.
L’analisi dei modelli di accumulo del virus avente diversità genomica durante un’epidemia può essere usata per fare inferenze sull’epidemiologia processi epidemiologici.
Questo è l’obiettivo di un corpo di tecniche filogenetiche che rientrano nell’ombrello termine filodinamica, coniato da Grenfell et al. (47).
I metodi filodinamici sono utili nelle indagini sui focolai, poiché possono integrare e aumentare altre analisi epidemiologiche basate su casi confermati identificati.
In primo luogo, diversi approcci filodinamici possono essere meno influenzati – o diversamente influenzati – da distorsioni nella diagnostica di sorveglianza, come i cambiamenti nello sforzo di sorveglianza nel tempo o l’individuazione disomogenea dei casi.
In secondo luogo, la filodinamica può rivelare caratteristiche dell’epidemia che si verificano al di fuori della finestra temporale di campionamento (per esempio, prima che venga identificato il primo caso).
In terzo luogo, le analisi filodinamiche forniscono un mezzo diretto per conoscere le dinamiche di popolazione di specifici lignaggi di virus diversi.
I metodi filodinamici utilizzano modelli probabilistici per legare l’albero filogenetico dei genomi campionati ai parametri epidemiologici di interesse. Come tali, richiedono l’inferenza di un albero filogenetico datato che contiene informazioni non solo su quali sequenze si raggruppano insieme, ma anche quando sono esistiti gli antenati comuni più recenti non campionati (MRCA) dei genomi virali campionati.
Mentre le date di campionamento sono note per i virus dai campioni sequenziati (cioè le punte dell’albero, vedi Fig. 1), gli MRCA (cioè i nodi interni) sono filogeneticamente dedotti e il loro tempo di esistenza deve essere stimato. La stima di queste date richiede l’uso di un modello di orologio molecolare parametrizzato da un tasso di clock – il tasso medio di sostituzione genetica lungo i rami della filogenesi.
Ci sono diverse famiglie distinte di modelli filodinamici: modelli coalescenti, nascita-morte e basati sulla simulazione. Recensioni di questi diversi modelli sono disponibili altrove (48, 49).
2.4 Caratteristiche genomiche ed evolutive di SARS-CoV-2 importanti per applicazioni genomiche
Diverse caratteristiche fondamentali di ogni virus determinano i possibili approcci per la generazione e l’uso dei dati genomici dei virus per informare le autorità sanitarie pubbliche.
Queste caratteristiche includono il suo materiale genetico (RNA o DNA), lunghezza genomica, struttura e composizione del genoma e tasso di evoluzione.
Il SARS-CoV-2 è classificato nel genere Betacoronavirus (sottogenere Sarbecovirus) nella famiglia Coronaviridae (sottofamiglia Orthocoronavirinae), una famiglia di virus a RNA positivo a singolo filamento RNA (50).
Il Comitato internazionale sulla tassonomia dei virus (ICTV) attualmente considera la SARS-CoV-2 come appartenente alla specie Severe acute respiratory syndrome-related coronavirus, insieme al SARS-CoV e ai virus strettamente correlati campionati da specie non umane specie non umane (51).
Il ceppo di riferimento di SARS-CoV-2, Wuhan-Hu-1 (accesso GenBank MN908947), è stato campionato da un paziente a Wuhan, in Cina, il 26 dicembre 2019 (52).
Questo genoma è 29 903 nucleotidi (nt) di lunghezza e comprende un ordine genico di struttura simile a quello visto in altri coronavirus: 5′-replicase ORF1ab-S-E-M-N-3′.
La replicasi predetta ORF1ab di Wuhan-Hu-1 è lungo 21 291 nt.
Si prevede che la poliproteina ORF1ab sia scisso in 16 proteine non strutturali.
ORF1ab è seguito da un certo numero di open reading frame (ORF) a valle.
Questi includono i predetti S (spike), ORF3a, E (envelope), M (membrana) e N (nucleocapside) di lunghezza 3822, 828, 228, 669 e 1260 nt, rispettivamente (52).
Come SARS-CoV, Wuhan-Hu-1 contiene anche un gene ORF8 previsto (366 nt di lunghezza) situato tra i geni M e N.
Infine, le sequenze terminali 5′ e 3′ di Wuhan-Hu-1 sono sono anch’esse tipiche dei betacoronavirus e hanno una lunghezza di 265 nt e 229 nt, rispettivamente.
Le stime preliminari del tasso evolutivo di SARS-CoV-2 sono vicine a una media di 1 x 10-3 sostituzioni per sito all’anno (45, 53), che è simile al tasso evolutivo medio osservato in altri genomi di virus a RNA (46).
Al momento in cui scriviamo, non esiste una stima accurata del tasso di mutazione per genoma replicazione per SARS-CoV-2 (tasso di mutazione). Tuttavia, ci si aspetta che sia simile a quello di altri coronavirus.
Il tasso di mutazione dei coronavirus e di altri membri dell’ordine dei Nidovirales è inferiore a quello di altri virus RNA perché hanno un’intrinseca capacità di proof-reading per correggere gli errori replicativi che è assente in altri virus a RNA (50).
3. Considerazioni pratiche per l’implementazione di un programma di sequenziamento genomico dei virus
Molti laboratori di salute pubblica ora riconoscono il potenziale impatto che le sequenze genomiche dei virus potrebbero avere sulle decisioni di salute pubblica durante l’attuale pandemia di COVID-19 o future epidemie (vedi anche la sezione 5).
3.1 Pianificazione di un programma di sequenziamento
I laboratori dovrebbero avere piani chiari in atto.
Una lista di controllo per aiutare la pianificazione è riportata nell’allegato 2.
Le domande chiave da considerare prima di iniziare un programma di sequenziamento includono le seguenti.
(1) Quali sono i risultati attesi dal programma di sequenziamento ?
(2) Quali campioni dovrebbero essere sequenziati per ottenere i risultati attesi identificati nella fase 1 ? Quali metadati o fonti di dati aggiuntivi sono critici ?
(3) Chi sono i principali stakeholder e quali sono le loro responsabilità ? Come possono essere efficacemente coinvolti ?
(4) Come si possono trasferire rapidamente e adeguatamente i campioni e le informazioni tra le parti interessate, come richiesto ?
(5) Il progetto è concepito in conformità con le leggi locali, nazionali e internazionali e con le linee guida etiche ?
(6) Sono disponibili finanziamenti adeguati, attrezzature e risorse umane per fornire tutte le fasi del recupero dei campioni, del sequenziamento in laboratorio, delle analisi bioinformatiche, filodinamiche e di altro tipo, della condivisione dei dati e della comunicazione di risultati tempestivi alle parti interessate ?
(7) Come si possono raggiungere gli obiettivi senza interrompere altre aree di lavoro del laboratorio, come la diagnostica clinica, ed evitando la duplicazione degli sforzi ?
(8) Come sarà valutato il programma per il rapporto costo-efficacia e l’impatto ?
3.2 Considerazioni etiche
Quando si progetta un programma di sequenziamento, è importante esaminare tutte le implicazioni etiche.
Devono essere identificati i possibili rischi di danno per i partecipanti alla ricerca e devono essere strategie di mitigazione dovrebbero essere definite.
Tutte le indagini proposte dovrebbero essere valutate e approvate da un comitato di revisione etica, tenendo conto del valore sociale e della validità scientifica dell’indagine dell’indagine, la selezione dei partecipanti, il rapporto rischio-beneficio, il consenso informato e il rispetto dei partecipanti (54, 55).
Quando i ricercatori hanno poca esperienza nell’identificare possibili problemi etici questioni etiche relative al sequenziamento di agenti patogeni, la collaborazione internazionale e l’impegno di competenze appropriate sono fortemente incoraggiati.
La collaborazione tra i ricercatori di tutto il mondo aiuterà a garantire partnership di ricerca eque e reciprocamente vantaggiose.
I ricercatori locali hanno maggiori probabilità di comprendere i loro sistemi di assistenza sanitaria e di ricerca e di essere in grado di tradurre i risultati in politiche, e quindi sono spesso più adatti ad assumere ruoli di guida e attivi in tutto il il processo di ricerca (54,55).
Le considerazioni etiche relative alla condivisione dei dati sono discusse più dettaglio nel capitolo 4.
3.3 Identificare i risultati attesi e i dati necessari
Prima di intraprendere qualsiasi programma di sequenziamento, dovrebbero essere stabiliti degli obiettivi realizzabili.
Gli obiettivi possibili sono discussi ampiamente nella sezione 5 ; gli obiettivi definiti influenzeranno la progettazione del flusso di lavoro di sequenziamento.
Una volta che gli obiettivi sono stati identificati, deve essere progettata una strategia di campionamento realizzabile per raccogliere le sequenze genomiche e i metadati appropriati ; le sequenze genomiche che mancano di metadati appropriati non sono utili per la maggior parte delle applicazioni.
Diverse domande di salute pubblica richiederanno diverse strategie di campionamento e dati.
È quindi di vitale importanza garantire che ci sia una discussione tra le diverse parti interessate che (a) conducono il campionamento diagnostico, (b) scelgono i campioni per il sequenziamento, (c) scelgono la strategia di sequenziamento, (d) scelgono le strategie analitiche e (e) utilizzano le informazioni generate per la salute pubblica, per garantire che le strategie di campionamento genomico e la raccolta di metadati siano correttamente mirate per le analisi a cui sono destinate.
3.4 Identificazione e collegamento con le parti interessate
Gli attori chiave dovrebbero essere identificati, consultati e coinvolti in una fase iniziale (Box 2).
La loro identità e il livello di coinvolgimento varieranno a seconda delle circostanze locali e degli obiettivi del programma, ma è ragionevole considerare gli stakeholder coinvolti in tutte le fasi del processo, dall’identificazione del caso all’uso dei risultati.
Può essere rilevante fornire risorse educative risorse educative alle parti interessate, compreso il pubblico in generale, per dimostrare la potenziale utilità di un programma di sequenziamento e per spiegare come le sequenze saranno utilizzate e perché sono necessari metadati specifici del paziente sono necessari.
Una stretta collaborazione e comunicazione tra le parti interessate sono critiche se le attività di sequenziamento devono risolvere questioni di importanza per la salute pubblica.
Box 2. Stakeholder da coinvolgere nello sviluppo di programmi di sequenziamento
Questo elenco non è esaustivo e dovrebbero essere considerati altri stakeholder, a seconda delle circostanze locali.
– Organismi di salute pubblica. Gli enti sanitari pubblici locali o nazionali, come i ministeri della sanità, spesso commissionano o aiutano a fornire programmi di sequenziamento della SARS-CoV-2.
Il loro coinvolgimento assicurerà che gli obiettivi rispondano a questioni politiche chiave. Inoltre, gli enti di salute pubblica Inoltre, gli enti sanitari pubblici possono spesso aiutare a garantire la raccolta diffusa di particolari campioni diagnostici e metadati.
– I laboratori diagnostici dovrebbero idealmente essere partner in qualsiasi programma di sequenziamento per SARS-CoV-2. Essi hanno tipicamente il miglior accesso ai campioni di SARS-CoV-2 e spesso possono fornire campioni positivi residui e metadati direttamente alle strutture di sequenziamento.
In alcuni impostazioni, i laboratori di diagnostica clinica possono essere incaricati di implementare un programma di programma di sequenziamento, mentre in altri il sequenziamento può essere effettuato da laboratori di ricerca esterni o da laboratori nazionali di salute pubblica.
–Le strutture di sequenziamento possono essere pubbliche o private ; alcune strutture di sequenziamento avranno la capacità bioinformatica per generare genomi di virus di consenso, mentre altri forniranno dati grezzi che devono essere ulteriormente elaborati altrove per generare genomi.
Non tutti i bioinformatici avranno l’esperienza per gestire i dati prodotti da tutte le possibili tecniche e piattaforme di tecniche e piattaforme di sequenziamento in laboratorio.
In questi casi, il supporto di un esperto che in grado di gestire il tipo di dati previsto è fortemente raccomandato.
– I gruppi analitici che condurranno analisi filogenetiche, filodinamiche o altre analisi genomiche devono essere strettamente coinvolti nel determinare quali campioni dovrebbero essere sequenziati, in modo che le sequenze genomiche siano appropriate per i metodi analitici da utilizzati.
Non si dovrebbe dare automaticamente per scontato che la competenza per condurre tali analisi sia presenti nei laboratori di genetica molecolare che effettuano il sequenziamento.
Ove pertinente, una stretta integrazione tra gli analisti e coloro che sono coinvolti nella sorveglianza e nella risposta (per esempio team di salute pubblica che indagano sui focolai locali) aumenterà l’impatto potenziale delle analisi.
– I team di prevenzione e controllo delle infezioni (ad esempio in ospedale, nelle case di riposo e nella sanità pubblica) possono sostenere l’identificazione di cluster di malattie emergenti e sono ben posizionati per identificare casi utili per il sequenziamento. Possono anche agire sui risultati successivi riguardanti i cluster di trasmissione.
– I servizi di salute occupazionale in contesti lavorativi possono aiutare a identificare potenziali cluster di trasmissione o vie di trasmissione che possono essere studiate utilizzando il virus genomico studi genomici, e ad implementare le attività di prevenzione e controllo delle infezioni che emergono dai risultati di questi studi.
– I pazienti dovrebbero essere coinvolti per garantire che capiscano come le sequenze e i metadati vengono utilizzati e condivisi, e beneficiare dei risultati.
Un programma di coinvolgimento della comunità adeguatamente progettato e dotato di risorse programma di coinvolgimento della comunità può aiutare a identificare e affrontare i potenziali ostacoli alla alla ricerca, relativi per esempio allo stigma, e assicurare che la progettazione del programma sia consapevole e risponda all’ambiente socioculturale in cui il programma sarà attuato.
Una volta identificati i principali stakeholder, è necessario stabilire adeguati canali di comunicazione tra i vari gruppi.
Come minimo, gli obiettivi del programma dovrebbero essere definiti in un quadro multidisciplinare che coinvolga i rappresentanti senior di tutte le parti interessate.
La comunicazione tra le parti interessate dovrebbe idealmente essere mantenuta per tutta la durata del progetto, e può richiedere incontri quotidiani o settimanali tra i rappresentanti di alcuni o di tutti gli enti coinvolti, per assicurare reazioni appropriate a situazioni mutevoli durante l’epidemia (per esempio, l’indagine di cluster di trasmissione non appena si presentano).
Attività focalizzate sull’epidemiologia che integrano gli analisti di dati genomici dati genomici direttamente nei team di investigazione e risposta della salute pubblica hanno probabilmente un maggiore impatto immediato rispetto a quelle in cui l’analisi genomica dei virus è considerata un’attività separata o secondaria. separata o secondaria.
Come, quando e con chi vengono condivisi i dati – con la comunità scientifica o tra parti interessate – dovrebbe essere concordato all’inizio.
Le responsabilità delle parti interessate, compresa la fornitura di finanziamento, se del caso, dovrebbero essere concordate.
Se saranno generati dati o pubblicazioni, è spesso utile concordare in anticipo il modo in cui le persone coinvolte saranno giustamente accreditate per il loro contributo alla produzione o analisi dei dati.
I risultati dell’analisi di sequenziamento dovrebbero essere comunicati rapidamente alle parti interessate in un relazione scritta standardizzata e facilmente interpretabile, e dovrebbero essere organizzate opportunità di discussione organizzate.
Il messaggio pratico dei risultati e dei limiti analitici dovrebbe essere essere trasmesso in un linguaggio quotidiano, evitando il gergo tecnico.
Dove un approccio multidisciplinare è stato seguito nel trattare questioni di salute pubblica (per esempio, coinvolgendo analisi di filogenetica e modellazione matematica), i risultati del sequenziamento dovrebbero idealmente essere discussi accanto ai risultati di altri campi.
3.5 Esecuzione del progetto : acquisizione di dati, logistica e risorse umane
Le considerazioni tecniche riguardanti l’aderenza legale ed etica, la selezione del campione, la valutazione dettagliata delle risorse
valutazione dettagliata delle risorse e la guida tecnica sono riportate nella sezione 6.
3.6 Valutazione del progetto
Un feedback strutturato regolare dovrebbe essere richiesto alle parti interessate per identificare e affrontare qualsiasi difficoltà che possano sorgere.
Il potenziale del sequenziamento genomico dei virus continua a crescere e la comunità scientifica e di salute pubblica comunità stanno rapidamente sviluppando nuove strategie per massimizzare il suo impatto nella futura malattia epidemie.
Tutti gli sforzi di sequenziamento dovrebbero quindi includere chiare opportunità per una regolare valutazione da parte di tutte le parti interessate di ciò che è stato utile, ciò che è mancato e quale impatto il sequenziamento raggiunto.
Identificare e comunicare questi risultati ai ricercatori e agli enti che li finanziano è importante per aiutare a guidare lo sviluppo di nuovi strumenti.
4. Condivisione dei dati
4.1 Raccomandazioni dell’OMS sulla condivisione dei dati
La rapida condivisione dei dati di sequenza del genoma dell’agente patogeno, insieme ai relativi metadati epidemiologici e clinici anonimizzati, massimizzerà l’impatto del sequenziamento genomico nella risposta di salute pubblica.
Tali dati, generati durante un’epidemia, dovrebbero essere condivisi con la comunità globale il più rapidamente possibile, per garantire la massima utilità nel migliorare la salute pubblica.
Nell’aprile 2016, l’OMS ha rilasciato una dichiarazione politica sulla condivisione dei dati nel contesto delle emergenze di salute pubblica :
“L’OMS sosterrà che le sequenze del genoma degli agenti patogeni siano rese pubblicamente disponibili il più rapidamente possibile attraverso le banche dati pertinenti e che i benefici derivanti dalla utilizzo di tali sequenze siano condivisi equamente con il paese da cui proviene la sequenza del genoma del patogeno ha origine” (56).
Uno dei fattori critici per assicurare la continua condivisione di dati genetici è il dovuto riconoscimento a coloro che raccolgono campioni clinici e generano sequenze del sequenze del genoma dei virus.
Le fonti dei dati dovrebbero essere riconosciute laddove i dati disponibili pubblicamente sono e le pubblicazioni correlate e gli articoli pre-print dovrebbero essere citati, se disponibili.
Inoltre, i finanziatori, editori di riviste e revisori di pari livello dovrebbero incoraggiare la condivisione dei dati.
4.2 Condivisione di metadati appropriati
I metadati dei campioni resi anonimi dovrebbero essere condivisi insieme ai dati genomici della SARS-CoV-2 per massimizzare l’utilità della sequenza genomica.
I metadati condivisi dovrebbero sempre includere almeno la data e il luogo di raccolta del campione, ma i metadati aggiuntivi aumenteranno notevolmente le potenziali applicazioni della sequenza.
Dove possibile, quindi, i metadati dovrebbero includere dati relativi al tipo di campione, come la sequenza è stata ottenuta, collegamenti ad altri virus sequenziati, la storia del viaggio del paziente e le informazioni demografiche o cliniche.
Per una descrizione dettagliata dei metadati si veda la sezione 6, Tabella 2.
Quando qualsiasi informazione viene condivisa, è importante che il l’anonimato del paziente sia protetto.
4.3 Condivisione di sequenze di consenso, sequenze parziali di consenso sequenze e dati di sequenza grezzi
Poiché il SARS-CoV-2 è emerso solo recentemente negli esseri umani, la diversità genetica del virus rimane relativamente limitata e le sequenze a lunghezza intera sono quindi importanti per catturare quanti più siti filogeneticamente informativi possibile.
Se il sequenziamento di tutta la lunghezza non ha successo, possono essere generate sequenze parziali.
I genomi di SARS-CoV-2 che hanno una copertura parziale sono ancora preziosi e dovrebbero essere condivisi.
Mentre la copertura del genoma richiesta (proporzione di siti senza basi ambigue, cioè Ns) varierà per diverse applicazioni e per diversi virus, i genomi parziali spesso rappresentano importanti fonti di dati.
Per esempio, i genomi del virus Zika con una copertura di appena il 40% (cioè il 60% dei siti con Ns) sono risultati filogeneticamente informativi della struttura del clade (57).
Come per i genomi completi, la qualità del genoma parziale dovrebbe essere controllata per garantire che i siti con supporto insufficiente siano mascherati prima che il genoma sia reso disponibile al pubblico.
Parziale genomi parziali in cui la copertura o la profondità di sequenziamento è generalmente molto bassa, ma in cui alcune brevi regioni hanno una profondità di sequenziamento molto alta, può essere indicativo di contaminazione con ampliconi prodotti attraverso la reazione a catena della polimerasi (PCR) e dovrebbero essere attentamente valutati prima di condividere.
La condivisione di letture di sequenziamento grezze (cioè tutti i singoli frammenti sequenziati di un genoma virale prima che vengano assemblati in un genoma di consenso) è importante perché permette l’effetto di diversi approcci bioinformatici per la generazione del genoma di consenso da confrontare direttamente e facilita la correzione degli errori, se necessario.
A seconda della strategia di sequenziamento adottata e la profondità della copertura di sequenziamento, i dati a livello di lettura possono anche essere utilizzati per le analisi di variazione intra-ospite nei genomi dei virus.
I set di dati a livello di lettura di SARS-CoV-2 dovrebbero quindi essere resi disponibili quando possibile.
Dato che la dimensione dei dati delle librerie sequenziate può raggiungere centinaia di gigabyte, la condivisione dei dati a livello di lettura può essere più difficile in ambienti che hanno limitate velocità di upload su internet o connessioni intermittenti.
I dati grezzi contenenti letture umane devono essere filtrati per mantenere solo dati di sequenze genetiche non umane (cioè virali) prima della condivisione, per fine di garantire l’anonimato del paziente (vedi sezione 6.7.1).
4.4 Piattaforme per la condivisione
La condivisione delle sequenze tramite piattaforme di ricerca comunemente usate aumenta l’accessibilità dei dati.
Le piattaforme variano nel tipo di dati che ospitano, le condizioni d’uso che pongono sui dati e la facilità con cui i metadati possono essere caricati. Alcune piattaforme (per esempio l’European Nucleotide Nucleotide Archive) offrono modelli di fogli di calcolo per i dati di sequenza che possono essere compilati offline e poi caricati in lotti.
I meccanismi di condivisione utilizzati per i dati di sequenze genomiche includono i database di dominio pubblico e ad accesso pubblico.
I database di dominio pubblico forniscono l’accesso ai dati senza richiedere l’identità di coloro che l’accesso e l’utilizzo dei dati.
Nei database ad accesso pubblico, gli utenti devono identificarsi per assicurare un uso trasparente dei dati e permettere una supervisione efficace, per proteggere i diritti di chi contribuisce ai dati contribuenti, fare del loro meglio per collaborare con i fornitori di dati e riconoscere il loro contributo nei risultati pubblicati. contributo nei risultati pubblicati.
Le sequenze genetiche della SARS-CoV-2 con i metadati appropriati sono frequentemente condivise attraverso piattaforme multiple.
I database di dominio pubblico per la condivisione dei genomi di consenso genomi di consenso includono il National Centre for Biotechnology Information (NCBI), l’European Molecular Biology Laboratory’s European Bioinformatics Institute (EMBL-EBI), e la DNA Data Bank of Japan (DDBJ).
I dati grezzi letti con metadati appropriati sono condivisibili tramite repository dell’International Nucleotide Sequence Data Collaboration (INSDC), che include il NCBI Sequence Read Archive (SRA), l’EMBL-EBI ENA e il DDBJ Archivio delle letture di sequenza.
Un database ad accesso pubblico per i genomi di consenso è per esempio GISAID EpiCoV™.
Il portale dati COVID-19 cerca di facilitare la condivisione e l’accesso a tutte le fonti di dati biomedici che sono rilevanti per COVID-19 (58).
I laboratori dovrebbero contattare le piattaforme di condivisione delle sequenze per aggiornare le sequenze parziali precedentemente inviate se viene identificato e corretto un errore.
Le analisi preliminari dei dati di sequenza genetica sono spesso condivise su forum e server di preprint, come medRxiv o bioRxiv.
Questo permette ai produttori di dati di fornire ulteriori informazioni aggiuntive sui risultati iniziali alla comunità scientifica più ampia.
I forum, tra cui Virological si sono dimostrati utili per la condivisione informale e la discussione dei risultati iniziali con la genetica molecolare, e i post possono essere continuamente aggiornati man mano che le analisi progrediscono.
I server di preprint sono spesso utilizzati per condividere articoli al punto di presentazione ad una rivista peer-reviewed, e chiaramente comunicare chiaramente le intenzioni di pubblicazione.
L’OMS incoraggia fortemente la condivisione di dati genetici e metadati il più presto possibile dopo i controlli di qualità dei dati, senza trattenerli fino a dopo il deposito di preprint deposizione.
Le analisi preliminari non revisionate vengono usate più estesamente dal pubblico e dai media nell’attuale pandemia che mai.
Gli scienziati dovrebbero quindi essere consapevoli di come le analisi potrebbero essere interpretate o presentate dai media, e dovrebbero fornire chiare interpretazioni dei loro risultati in modo che i risultati non possano essere facilmente fraintesi.
5. Applicazioni della genomica alla SARS-CoV-2
Questa sezione esamina come il sequenziamento del genoma della SARS-CoV-2 è stato utilizzato nelle diverse fasi della della pandemia COVID-19 e suggerisce possibili applicazioni future.
Fornisce anche una breve indicazioni sui limiti comuni degli approcci attuali, per aiutare a fissare obiettivi realistici.
Per alcune delle applicazioni considerate, il sequenziamento genomico del virus rappresenta solo una piccola componente di un’indagine più ampia, che può includere un laboratorio essenziale sostanziale o indagini cliniche.
5.1 Capire l’emergenza della SARS-CoV-2
5.1.1 Identificazione dell’agente causale del COVID-19
Il SARS-CoV-2 è stato indipendentemente identificato e sequenziato all’inizio del 2020 da Wu et al., Lu et al. e Zhou et al. (52, 59,60).
Diversi approcci metagenomici di sequenziamento di nuova generazione (mNGS) sono stati utilizzati per identificare il patogeno causale di COVID-19.
Il sequenziamento metagenomico permette il sequenziamento non mirato dell’acido nucleico in un campione e può quindi identificare RNA o DNA virale se presente con un numero di copie abbastanza alto rispetto al DNA o RNA di altre fonti fonti (vedi anche sezione 6.5.1).
Il completamento delle sequenze complete del genoma del virus, compresi i termini del genoma, generalmente ha coinvolto il sequenziamento Sanger e un’amplificazione rapida 5’/3′ delle cDNA ends (RACE).
Questo metodo è efficiente in termini di costi per il sequenziamento di brevi regioni di un genoma che può essere perso con i metodi metagenomici, ma si basa sulla conoscenza precedente delle informazioni sulla sequenza relativamente vicina alla regione mancante.
5.1.2 Determinazione dei tempi di origine e di diversificazione precoce
Era particolarmente importante determinare quando la SARS-CoV-2 è emersa per la prima volta negli esseri umani, poiché questo potrebbe fornire un’indicazione sul fatto che ci sia stato un lungo periodo di trasmissione non rilevata prima che si vedessero i primi casi clinici (e quindi forse molti casi non rilevati).
I genomi del CoV-2 di Wuhan e delle aree circostanti della provincia di Hubei hanno fornito una serie di intuizioni chiave.
Tutte le sequenze erano estremamente correlate, differendo solo per poche varianti nucleotidiche.
Diversi primi esercizi di datazione dell’orologio molecolare utilizzando queste sequenze hanno dato tempi stimati per la comparsa dell’antenato comune più recente di tutti i virus SARS-CoV-2 sequenziati come il periodo da novembre a dicembre 2019.
Queste stime iniziali sono state confermate man mano che più sequenze sono diventate disponibili.
L’ultima data possibile di comparsa della SARS-CoV-2 nell’uomo è quindi novembre-dicembre 2019.
Questo è vicino alla prima identificazione del cluster iniziale di casi di polmonite a Wuhan a metà dicembre (59-61).
Quando si è verificata una sola introduzione nell’uomo, la prima tempistica possibile dell’emergenza di un virus zoonotico nell’uomo è filogeneticamente rappresentata dal tempo all’antenato comune più recente (TMRCA) del virus zoonotico umano e del virus animale non umano da cui è emerso.
Il campionamento inadeguato dei virus animali non umani che sono strettamente correlati alla SARS-CoV-2 significa che il possibile intervallo in cui la SARS-CoV-2 potrebbe essere emersa negli nell’uomo è relativamente ampio se si considerano solo i dati filogenetici.
È quindi difficile distinguere filogeneticamente tra due possibili scenari di comparsa della SARS-CoV-2.
In primo, la SARS-CoV-2 potrebbe essere emersa nell’uomo alla fine del 2019, vicino al momento dell’identificazione della malattia identificazione.
In alternativa, un progenitore della SARS-CoV-2 potrebbe essere emerso e aver circolato negli prima di acquisire i cambiamenti genomici che gli hanno permesso di causare un gran numero di casi gravi e dare inizio all’attuale pandemia (62).
Tuttavia, nessun campione raccolto da esseri umani prima della fine del 2019 è stato ancora trovato positivo per la SARS-CoV-2 ; il secondo possibile scenario è, quindi, attualmente non è supportato da altre linee di prova.
Sebbene le sequenze di Wuhan mostrino una diversità genetica limitata, sono evidenti due linee filogeneticamente distinte sono evidenti, indicando un evento di separazione all’inizio della comparsa del virus.
Si noti che distinzione filogenetica dei lignaggi non implica differenze fenotipiche nella trasmissibilità o patogenicità tra i lignaggi, perché tali distinzioni di solito emergono attraverso stocastico processi stocastici.
Questi lignaggi sono stati recentemente classificati come lignaggi A e B (61) (più raramente riferiti come lignaggi S e L) (vedere la sezione 6.8.7 per ulteriori discussioni sulla nomenclatura dei lignaggi della SARS-CoV-2).
In particolare, anche se i virus del lineage B sono stati identificati e sequenziati per primi (52, 59, 60), è probabile che i virus del lignaggio A siano ancestrali perché condividono due nucleotidi con i coronavirus più strettamente correlati in altri animali che non sono condivisi nei virus di discendenza B.
Nonostante le forti misure di quarantena adottate nella provincia di Hubei, entrambi i lignaggi sono stati esportati nel resto della Cina e hanno seminato molteplici epidemie in altri paesi.
5.1.3 Identificare l’origine zoonotica
Le sequenze del genoma della SARS-CoV-2 e i genomi dei virus correlati di altri animali sono stati analizzati filogeneticamente nel tentativo di determinare il serbatoio zoonotico da cui la SARS-CoV-2 è emersa.
Fino ad oggi, c’è stato un campionamento relativamente limitato con lo scopo di identificare gli animali coinvolti nella genesi della SARS-CoV-2 e determinare quando, dove e come il virus è emerso negli esseri umani.
Sebbene al mercato all’ingrosso dei frutti di mare di Huanan a Wuhan siano stati prelevati campioni ambientali al momento della sua chiusura all’inizio di gennaio 2020 (63) e siano risultati positivi, al momento non è chiaro se questi campioni provenissero solo dalle superfici o anche dagli animali presenti nel mercato.
Se il primo caso, questi potrebbero semplicemente riflettere la contaminazione umana.
Inoltre, non tutti i primi casi potrebbero essere collegati a questo mercato (61).
L’identificazione della fonte animale da cui è emersa la SARS-CoV-2 è emersa potrebbe aiutare a combattere la diffusione di teorie cospirative relative alla comparsa.
Le ricerche precedenti alla pandemia di COVID-19 hanno dimostrato che i betacoronavirus sono presenti in un numerose specie di mammiferi e presentano una diversità filogenetica particolarmente elevata nei pipistrelli (64- 66).
Che i pipistrelli abbiano probabilmente giocato un ruolo nella storia evolutiva della SARS-CoV-2 è stato confermato dall’identificazione di un parente stretto del SARS-CoV-2 (denominato RaTG13) in una specie di pipistrello a ferro di cavallo (Rhinolophus affinis) campionato nella provincia di Yunnan, in Cina, nel 2013 (60).
RaTG13 e SARS-CoV-2 hanno circa il 96% di somiglianza di sequenza in tutto il genoma anche se questo non esclude decenni di divergenza evolutiva tra loro (67).
Un altro coronavirus, RmYN02, è stato identificato in una specie diversa di pipistrello a ferro di cavallo, Rhinolophus malayanus, nella provincia di Yunnan nel 2019 (68).
Sebbene il genoma di RmYN02 abbia sperimentato una complessa serie di eventi di ricombinazione, è il parente più prossimo di SARS-CoV-2, condividendo una somiglianza di sequenza nucleotidica del 97% nel gene ORF1ab.
Parenti stretti del SARS-CoV-2 sono stati trovati anche nei pangolini malesi (Manis javanica) recuperati in attività di contrabbando nelle province di Guangdong e Guangxi nella Cina meridionale.
I coronavirus dei pangolini sono più distanti dalla SARS-CoV-2 rispetto a RaTG13 e RmYN02 nei loro genomi nel loro insieme, ma condividono una forte somiglianza di sequenza con SARS- CoV-2 nel dominio chiave di legame al recettore (RBD) del gene spike (S) (97,4% a livello di aminoacidi) (69). livello degli aminoacidi) (69).
Mentre è chiaro che i betacoronavirus sperimentano eventi di ricombinazione frequenti e complessi, e che questo processo si è verificato in virus che sono strettamente correlati alla SARS-CoV-2, non c’è prova al momento che la ricombinazione abbia giocato un ruolo diretto nell’emergere di questo virus (67).
Limitazioni. Anche se la SARS-CoV-2 ha senza dubbio origini animali, come la SARS-CoV e la MERS-CoV (64), la specie di origine sarà risolta solo con un ulteriore campionamento di un’ampia gamma di animali non umani.
È possibile che le sue origini non saranno mai completamente risolte.
5.2 Comprendere la biologia della SARS-CoV-2
5.2.1 Uso dei recettori dell’ospite
Poiché i virus possono replicarsi solo all’interno delle cellule viventi di un organismo ospite, determinare il recettore cellulare dell’ospite usato dalla SARS-CoV-2 è essenziale per comprendere la sua biologia di base.
Il legame del recettore è mediato dalla proteina S del virus.
Le somiglianze genetiche nel motivo di legame della proteina S tra la SARS-CoV-2 e altri coronavirus precedentemente studiati hanno aiutato a identificare il recettore cellulare a cui si lega la SARS-CoV-2 e quindi i tipi di cellule che potrebbe infettare.
Gli studi iniziali hanno indicato che il SARS-CoV-2 probabilmente utilizza lo stesso recettore cellulare dell’enzima di conversione dell’angiotensina 2 (ACE2) del SARS-CoV 2002-2003, ed è probabile che si leghi a questo recettore con alta affinità (70, 71).
La maggior parte dei residui aminoacidici che sono noti per essere essenziali per il legame di ACE2 da parte di SARS-CoV sono conservati in SARS-CoV-2 (70).
I saggi in vitro confermano la forte specificità per ACE2 suggerita dagli studi strutturali diretti (72).
Limitazioni. Sono stati necessari esperimenti in vitro o in vivo per la piena conferma dei risultati della sequenza genetica e sono sempre necessari per indagare qualsiasi cambiamento proposto nell’affinità di legame.(N.d.T. : in poche parole, hanno prima determinato il risultato, e poi hanno cercato come trovarlo, geniali…)
5.2.2 Evoluzione di SARS-CoV-2 : identificazione dei siti genomici candidati che possono conferire cambiamenti fenotipici
Tutti i virus acquisiscono cambiamenti genetici durante la loro evoluzione, e la maggior parte dei cambiamenti genetici acquisiti non influenzano sostanzialmente la virulenza o la trasmissibilità.
Non si può presumere che le varianti tra i genomi dei virus campionati da luoghi diversi siano la causa delle differenze epidemiologiche osservate in COVID-19 tra questi luoghi e sono invece probabilmente stocastiche.
Nonostante ciò, è possibile che si verifichi un cambiamento genetico che causa un corrispondente cambiamento fenotipico nella SARS-CoV-2 di importanza per la salute pubblica.
Studi genomici clinici adeguatamente condotti potrebbero essere utilizzati per proporre varianti candidate che potrebbero conferire cambiamenti fenotipici del virus clinicamente osservati, ma studi in vitro o in vivo dovrebbero essere condotti successivamente per valutare le varianti candidate.
Il sequenziamento genomico del virus prima e dopo tali studi sperimentali sarebbe anche necessario per escludere la possibilità che la differenza fenotipica dedotta non sia guidata da adattamenti stocastici del virus alla replicazione all’interno della coltura cellulare.
I fenotipi osservati nella cultura cellulare e nei modelli animali possono non tradursi in alterazioni nella malattia umana.
Quando i virus associati a diversi fenotipi hanno diversi siti che differiscono tra i genomi, può essere difficile determinare quali, se ce ne sono, di quelle varianti genetiche causano la differenza fenotipica osservata.
Le varianti genomiche identificate potrebbero essere studiate con la reverse genetica inversa per ottenere una comprensione completa delle loro caratteristiche fenotipiche.
La genetica inversa può coinvolgere l’induzione sintetica sistematica di un cambiamento genetico in un gene virale e lo studio dell effetto fenotipico che provoca in seguito alla produzione di quella proteina.
Tali esperimenti dovrebbero essere intrapresi solo in stretta osservanza delle leggi e dei regolamenti locali e (inter)nazionali sulla biosicurezza. leggi e regolamenti sulla biosicurezza.
Se un cambiamento genetico con un effetto fenotipico può essere confermato attraverso questi metodi, studi epidemiologici filodinamici (sezione 5.4) possono essere usati per tracciare la loro diffusione globale o locale diffusione.
Limitazioni. È estremamente impegnativo identificare e fornire prove dei cambiamenti genomici che possono conferire cambiamenti fenotipici.
Il sequenziamento genomico del virus è una parte necessaria di tali studi, che dovrebbero essere attentamente progettati e controllati al fine di convalidare qualsiasi ipotesi effetti ipotizzati.
Successivi studi in vitro e in vivo con virus mutanti possono, in alcuni casi, supportare ulteriormente le valutazioni di queste ipotesi.
5.3 Migliorare la diagnostica e la terapeutica
5.3.1 Migliorare la diagnostica molecolare
Mentre la SARS-CoV-2 è stata identificata per la prima volta nei pazienti attraverso il sequenziamento metagenomico (sezione 5.1), questo approccio è troppo lungo e costoso per essere usato di routine per diagnosticare l’infezione virale.
Lo sviluppo di test di amplificazione dell’acido nucleico rapidi, poco costosi e sensibili (NAAT) per il rilevamento molecolare di routine della SARS-CoV-2 è stato quindi considerato prioritario all’inizio dell’epidemia.
Il rapido rilascio pubblico dei genomi della SARS-CoV-2 è stato importante per la progettazione delle NAAT.
In particolare, questi genomi erano necessari per la progettazione di primer e sonde che si sarebbero legati efficacemente all’acido nucleico della SARS-CoV-2 (attraverso sequenze complementari esatte o quasi esatte) ma che non si legassero ad altri virus comunemente in circolazione, come i coronavirus che causano raffreddori comuni (73).
Molteplici NAAT SARS-CoV-2 sono state progettate e validate da diversi gruppi entro pochi giorni dal rilascio del primo genoma (ad esempio, 74-76).
Poiché la SARS-CoV-2 continua ad acquisire cambiamenti genetici nel tempo durante questa pandemia, la continua generazione e condivisione dei genomi del virus sarà vitale per monitorare la sensibilità prevista dei vari test diagnostici in luoghi diversi.
I disallineamenti tra i primer o le sonde e i corrispondenti siti di legame all’interno dei genomi di SARS-CoV-2 potrebbero ridurre la sensibilità del NAAT o provocare falsi negativi.
Il monitoraggio sarà particolarmente importante se un sito di variante viene rilevato in virus che sono filogeneticamente vicini.
L’utilizzo di obiettivi multipli per il rilevamento della SARS-CoV-2, come una PCR multiplex mirata a due o più regioni del genoma del virus, è un approccio conveniente per ridurre la possibilità di falsi negativi a causa dell’evoluzione del virus.
Il fallimento consistente nel rilevare un bersaglio in diversi campioni clinici, o l’emergere di differenze nella sensibilità dei test mirati a regioni diverse che non sono state osservate precedentemente e che si verificano in campioni clinici ma non nel controllo positivo stabilito, potrebbero essere
seguito dal sequenziamento del genoma del virus o del gene bersaglio per identificare la possibile causa.
Diverse piattaforme esistenti permettono di monitorare le discordanze tra le sequenze inviate dall’utente o quelle disponibili pubblicamente SARS-CoV-2 e i siti di legame primer/sonda dei NAAT comunemente usati.
Un certo numero di strumenti sono stati sviluppati per monitorare tali mismatch con i comuni comuni, come descritto altrove (77).
Limitazioni. Il sequenziamento genetico delle regioni di legame tra primer e sonda è sufficiente per studiare l’emergenza dei mismatch.
Tuttavia, il sequenziamento dell’intero genoma permette un’indagine genomica più ampia della diffusione spazio-temporale dei virus contenenti mismatch (ad esempio per determinare quando la variante di mismatch può essere sorta) o il numero di volte in cui la variante può essere emersa indipendentemente.
5.3.2 Supporto alla progettazione e al monitoraggio della sensibilità dei test sierologici
I dati sulla sequenza genomica del virus possono essere importanti per aiutare a identificare le proteine del virus che probabilmente essere fortemente antigeniche, e per indicare come questi antigeni possono essere prodotti per test sierologici test sierologici.
Lo screening peptidico ha indicato che le quattro proteine strutturali della SARS-CoV-2, S, E, M e N, sono probabilmente le più fortemente antigeniche (78, 79).
Gli antigeni della SARS-CoV-2 possono essere prodotti sinteticamente per l’uso in saggi commerciali.
In particolare, i geni sintetici del coronavirus che codificano le quattro proteine possono essere inseriti in sistemi di vettori di espressione (80, 81), dove le proteine sono prodotte.
Questo processo si basa sulla comprensione della sequenza genomica e della struttura delle proteine della SARS-CoV-2.
Man mano che la SARS-CoV-2 acquisisce sostituzioni genomiche, è possibile che emerga una stirpe con proprietà antigeniche alterate (sezione 5.2.2). Questo potrebbe significare che i test sierologici non riescono a rilevare che un individuo è stato infettato, perché l’antigene usato nel test è diverso da quello a cui l’individuo è stato esposto.
Inoltre, i test di rilevamento dell’antigene possono essere influenzati dal cambiamento virale, poiché gli anticorpi di cattura potrebbero non riconoscere la proteina virale adattata che mira a rilevare.
La valutazione continua della diversità genomica, anche nei siti antigenicamente importanti che possono essere sotto selezione, potrebbe aiutare a identificare siti candidati plausibili che potrebbero influenzare l’efficacia efficacia dei test sierologici.
Limitazioni. Le previsioni in silico del cambiamento antigenico dai dati di sequenza genomica sono inadeguate, e la possibile sensibilità dei test sierologici nel rilevamento di infezioni geneticamente infezioni geneticamente diverse dovrebbe sempre essere studiata attraverso la convalida sierologica di laboratorio.
5.3.3 Supporto alla progettazione del vaccino
Sono stati progettati diversi candidati vaccini contro la SARS-CoV-2 e alcuni sono stati valutati clinicamente (82).
Le sequenze del genoma della SARS-CoV-2 sono state utilizzate nella progettazione di candidati vaccini che si basano sull’inoculazione di antigeni o mRNA/DNA per stimolare, direttamente o indirettamente, la produzione di anticorpi e le risposte cellulari.
Molti dei primi candidati vaccini mRNA sono stati progettati esclusivamente sulla base dei genomi di SARS-CoV-2 pubblicamente disponibili.
In alternativa, i genomi sintetici dei coronavirus possono essere inseriti in sistemi di vettori di espressione (80, 81) per produrre antigeni per i vaccini (sezione 5.3.2).
Limitazioni. Mentre le sequenze genomiche possono aiutare nella progettazione di vaccini candidati, in vivo studi in vivo e studi clinici rimangono critici per valutare l’efficacia del vaccino.
5.3.4 Supporto alla progettazione della terapia antivirale
Lo sviluppo di nuovi farmaci antivirali può richiedere molto tempo.
La riproposizione di farmaci esistenti per il trattamento della SARS-CoV-2 potrebbe ridurre significativamente il tempo necessario per ottenere l’approvazione per l’uso clinico.
Le informazioni genetiche e strutturali possono rivelare somiglianze nei percorsi proteolitici e replicazione (78, 79) tra la SARS-CoV-2 e altri virus per i quali è già disponibile una terapia antivirale antivirali sono già disponibili, e quindi aiutano a determinare quali antivirali esistenti potrebbero essere
riproporre.
Diversi farmaci candidati che hanno come bersaglio proteine virali simili a quelle della SARS-CoV-2 sono già stati identificati (83) e sono attualmente oggetto di studi preclinici e clinici.
5.3.5 Identificare le mutazioni di resistenza antivirale o di non efficacia dal vaccino
Una volta che i vaccini sono implementati e/o gli antivirali diventano disponibili, il sequenziamento genomico potrebbe essere usato per supportare la sorveglianza delle varianti che possono conferire resistenza antivirale o permettere al vaccino di non essere efficace.
Il sequenziamento genomico o genetico approfondito può essere utile per esplorare l’impatto della diversità intra- ospite sulla resistenza antivirale e sulla non efficacia dal vaccino (se queste si verificano) o sulla patogenesi.
Un sequenziamento genetico di regioni specifiche di interesse, come il gene spike, può essere sufficiente per valutare la prevalenza di specifiche varianti note in regioni pre-identificate.
Limitazioni. Tali studi sono estremamente complessi e richiederanno un’indagine genomica mirata e dettagliata e l’indagine computazionale di virus da pazienti con una storia di vaccinazione nota e risultati clinici.
Mentre i dati di sequenza dei virus coltivati sotto pressione di selezione dei farmaci possono rivelare possibili marcatori di resistenza antivirale, questi marcatori dovrebbero sempre essere convalidati dalla reverse genetica inversa per determinare le loro caratteristiche fenotipiche.
5.4 Indagini sulla trasmissione e la diffusione del virus
Il posizionamento delle sequenze all’interno di un albero filogenetico può essere utilizzato per indagare le ipotesi di vie di trasmissione.
Il raggruppamento filogenetico di sequenze provenienti da pazienti esposti alla stessa ipotetica fonte di esposizione sarebbe coerente con (anche se non una forte evidenza per) quell’esposizione.
Sequenziando una parte dei casi al di fuori di un cluster ipotizzato, e includendo sequenze di riferimento globali che sono geneticamente più vicine alle sequenze del cluster (per rappresentare il sfondo della diversità genomica), può aiutare a valutare la probabilità che le sequenze di un cluster filogenetico identificato con un legame epidemiologico ipotizzato siano raggruppate insieme per caso.
Più alta è la proporzione di virus che sono sequenziati dallo stesso tempo e luogo dei virus di interesse, ma che non sono identificati come probabilmente parte di quel cluster, più bassa è la possibilità che quelle sequenze di virus cadano in un cluster per caso.
Al contrario, una considerevole separazione filogenetica delle sequenze di virus provenienti da due pazienti (ad esempio la collocazione all’interno di diversi lignaggi ben supportati) indicherebbe che i due pazienti hanno acquisito infezioni da fonti diverse.
Il clustering filogenetico è stato ampiamente utilizzato per studiare le fonti di trasmissione e eventi di esposizione per la SARS-CoV-2.
In uno dei primi studi, è stato suggerito che il raggruppamento di delle sequenze dei pazienti sulla nave da crociera Grand Princess era coerente con un’unica introduzione del virus su quella nave, seguita da una trasmissione tra i passeggeri (84).
Il osservazione di cladi monofiletici di virus campionati da membri della stessa famiglia è coerente con la trasmissione diretta tra membri della famiglia, o l’infezione dalla stessa fonte (non campionata).
Le analisi dei cluster di trasmissione possono guidare le decisioni sull’eventuale necessità di ulteriori misure di controllo aggiuntive sono necessarie per prevenire la trasmissione futura in ambienti identificati.
Limitazioni. Le informazioni filogenetiche non possono essere utilizzate per confermare la trasmissione diretta del virus tra due pazienti o la trasmissione da una singola fonte a più pazienti, perché non si può escludere il coinvolgimento di altri individui o fonti di esposizione che non sono stati campionati.
Il tasso evolutivo della SARS-CoV-2 significa che le sostituzioni avvengono in media a un ritmo più lento rispetto alle trasmissioni tra pazienti, e quindi questo rimane vero anche se le sequenze enomiche campionate sequenze genomiche campionate sono identiche.
5.4.2 Identificare e quantificare i periodi di trasmissione
Una volta che c’è sufficiente diversità genetica all’interno di una stirpe di virus, il tasso di cambiamento evolutivo (tasso di sostituzione) può essere stimato (sezione 2.4).
Se il tasso di sostituzione può essere stimato, la diversità genetica diversità genetica tra due virus campionati con date di campionamento note può essere utilizzata per stimare il TMRCA.
Il TMRCA di un gruppo di virus fornisce una stima del limite inferiore della durata della della sua circolazione all’interno della popolazione campionata.
Fondamentalmente, la durata stimata della circolazione può precedere la prima identificazione clinica di un caso di settimane o mesi.
Approcci filogenetici sono particolarmente utili per identificare dove la circolazione non rilevata (o criptica) circolazione può essersi verificata, e nella stima delle possibili date di eventi non osservati.
Le analisi iniziali suggeriscono che la SARS-CoV-2 ha ora acquisito una diversità genetica sufficiente per consentire di applicare tali approcci di orologio molecolare (45, 59, 85).
Di conseguenza, sono stati utilizzati per stimare che il lignaggio pandemico di SARS-CoV-2 sia emerso nell’uomo entro novembre-dicembre 2019 al più tardi (53, 59, 85). (sezione 5.1.2).
Applicazioni importanti di questi approcci per il controllo della COVID-19 includono l’identificazione della trasmissione locale non rilevata in diverse località.
L’identificazione di una trasmissione locale di lunga durata, in gran parte non rilevata clinicamente può suggerire che luoghi o popolazioni specifici dovrebbero essere presi di mira con programmi di sorveglianza diagnostica più estesi o adattati.
Limitazioni. La risoluzione temporale degli eventi che possono essere studiati è limitata dal rapporto tra il tasso evolutivo e il tasso di trasmissione.
Le stime attuali del tasso evolutivo della SARS-CoV-2 sono che, in media, si verifica una sostituzione ogni 2 settimane circa.
Questo significa che gli eventi di trasmissione tra individui spesso non saranno risolvibili genomicamente, e gli eventi epidemiologicamente rilevanti che si verificano su una scala temporale più sottile non possono essere studiati con queste tecniche.
All’inizio dell’epidemia, è stato difficile stimare la durata della trasmissione criptica perché la SARS-CoV-2 non aveva ancora accumulato una diversità genomica sufficiente.
Pertanto, era difficile determinare se un particolare genoma fosse il risultato di una trasmissione locale o di una nuova introduzione da un luogo con una diversità circolante simile.
Gli studi hanno suggerito che il SARS-CoV-2 potrebbe aver circolato senza essere stato individuato per settimane a Seattle (USA) e in Italia prima del rilevamento clinico dei primi casi acquisiti in comunità (84, 86).
Tuttavia, uno studio successivo ha sostenuto che la durata della trasmissione criptica potrebbe essere stata sovrastimata di diverse settimane (87).
Errori nel sequenziamento o nella generazione del consenso possono oscurare i segnali filogenetici quando la vera diversità è bassa.
Gli errori di sequenziamento possono anche influenzare le stime della variazione del tasso evolutivo tra i lignaggi e i tempi di divergenza stimati.
La durata minima della trasmissione del virus può essere stimata anche quando sono molto pochi (due o più) casi di una singola catena di trasmissione sono sequenziati.
Tuttavia, incorporando ulteriori campioni aggiuntivi da un’ampia area geografica e da un periodo di tempo ridotto il rischio che i casi campionati si raggruppino strettamente all’interno di una filogenesi per caso, così che la durata minima stimata sarà probabilmente essere più vicina alla durata reale.
5.4.3 Identificare gli eventi di importazione e la circolazione locale
Se sono disponibili metadati sulla localizzazione del campionamento, il sequenziamento dei genomi della SARS-CoV-2 può aiutare a determinare se le infezioni sono derivate dalla trasmissione locale o sono state importate.
Tali dinamiche di trasmissione possono essere interpretate in modo cauto e informale attraverso il posizionamento delle sequenze all’interno di una filogenesi (Fig. 2) o studiate attraverso analisi filogeografiche più formali o analisi dei tratti discreti, in cui la posizione di ogni nodo interno della filogenesi viene stimata statisticamente.
L’incorporazione di tempi di campionamento noti permette di ricostruire il movimento spazio-temporale del focolaio.
L’inferenza filogeografica formale include sia approcci discreti che continui.
Nel primo, i lignaggi del virus sono considerati in movimento tra un numero fisso di luoghi distinti (88).
Le aree esatte sono definite dall’utente e possono rappresentare paesi, unità amministrative, città, ecc., a seconda delle domande specifiche poste. Nell’approccio continuo, il movimento del lignaggio del virus movimento del virus è modellato sulla base di processi di diffusione e camminata casuale tra coordinate geografiche (89).
Entrambe le indagini filogeografiche discrete e continue possono essere condotte sotto un certo numero di quadri statistici, che hanno diversi vantaggi e sfide; questi sono stati ampiamente rivisti altrove (90, 91).
Poiché la diversità genomica della SARS-CoV-2 era bassa durante i primi mesi della pandemia, l’uso del sequenziamento del genoma per tracciare la sua diffusione era in gran parte limitato alle introduzioni nazionali e introduzioni regionali, piuttosto che alla trasmissione nelle comunità.
L’interpretazione informale e visiva di strutture filogenetiche è stata utilizzata ampiamente nella letteratura iniziale per dedurre il movimento internazionale o movimento regionale.
Per esempio, l’epidemiologia genomica è stata usata per mostrare che molti casi sequenziati casi in Connecticut (USA) sono stati probabilmente importati attraverso viaggi interni da altre parti degli USA piuttosto che da altri paesi (92).
La valutazione filogeografica della diversità genomica potrebbe essere usata per valutare se per esempio una quarantena più rigorosa dei pazienti che hanno visitato luoghi specifici stia prevenendo efficacemente l’introduzione o l’esportazione della SARS-CoV-2 in altre regioni.
Per esempio, in Brasile, le analisi filogeografiche continue analisi filogeografiche continue hanno mostrato che la diffusione della SARS-CoV-2 all’interno e tra gli stati brasiliani è diminuita dopo l’implementazione di interventi non farmacologici (53).
Gli approcci filogeografici che incorporano anche i tempi di campionamento permettono di stimare sia dove e quando possono essersi verificati gli eventi di movimento del lignaggio del virus.
La durata della persistenza del virus persistenza del virus, il numero di introduzioni e la dimensione relativa del focolaio possono essere determinati per ogni luogo e possono quindi essere usati per identificare luoghi specifici in cui le misure di controllo devono essere rafforzate.
Può essere utile studiare i virus nei viaggiatori di ritorno per aiutare a ricostruire l’epidemiologia della SARS-CoV-2 nel paese in cui l’infezione è stata contratta (93).
I nuovi approcci permettono la storia dei viaggi dei pazienti e le sequenze provenienti da località non campionate per essere incorporate in analisi filogeografiche discrete, permettendo così di rivelare modelli filogeografici più realistici rivelare e valutare l’effetto del campionamento globale distorto (94).
Limitazioni. Le ricostruzioni filogeografiche sono spesso impegnative dal punto di vista computazionale.
Strategie di sottocampionamento attentamente considerate possono aiutare a ridurre questo carico computazionale (sezione 6.8.1).
La dispersione degli agenti patogeni umani non è sempre ben catturata da questi processi.
Tuttavia, laddove le scale geografiche e i viaggi a lunga distanza sono limitati, le passeggiate casuali possono adeguatamente catturare il movimento della SARS-CoV-2.
Dovrebbe essere data un’attenta considerazione all’adeguatezza di un processo continuo per la SARS-CoV-2, poiché l’uso di modelli di diffusione inappropriati può portare a conclusioni a conclusioni errate.
Il modo in cui le sequenze del genoma del virus vengono campionate può influenzare fortemente le conclusioni delle analisi filogeografiche.
È quindi estremamente importante condurre analisi e interpretare risultati con cautela, coinvolgendo idealmente esperti che hanno esperienza in questi metodi.
Ci sono molteplici modi in cui le analisi filogeografiche possono non riuscire a catturare i “veri modelli” di diffusione, compresi i seguenti :
-Il sottocampionamento dei genomi dei virus può portare a sottostimare il numero di introduzioni (e quindi sovrastimare l’estensione della trasmissione comunitaria) (Fig. 2).
Questo è stato chiaramente evidenziato da Lu et al. (59), che hanno dimostrato che un singolo cluster di sequenze strettamente correlate sequenze campionate da pazienti in Guangdong in realtà rappresentava più indipendenti introduzioni indipendenti attraverso i viaggi.
La storia dei viaggi dei pazienti è un’informazione importante che dovrebbe essere utilizzato per supportare i risultati filogenetici dove possibile (appropriata condivisione dei dati e protezione dell’anonimato dei pazienti è discussa nella sezione 4).
-Per le analisi filogeografiche discrete, la localizzazione dei virus ancestrali può essere dedotta in modo affidabile solo solo dall’insieme dei luoghi in cui sono stati osservati i virus campionati (89, 95).
Di conseguenza, con i soli dati genomici, è solitamente impossibile distinguere tra trasmissione diretta trasmissione tra due luoghi e la trasmissione indiretta attraverso un luogo intermedio in cui non sono stati prodotti genomi.
Distinguere tra questi scenari è possibile solo possibile solo in rare situazioni in cui le informazioni sul viaggio sono note (94).
-Per alcune analisi filogeografiche discrete (in particolare quelle basate sull’analisi discreta dei tratti piuttosto che su modelli di coalescenza o di nascita-morte), le località che hanno un numero maggiore di sequenze genomiche associate ad esse hanno maggiori probabilità di essere ricostruite come località donatrici dalle quali un virus si diffonde successivamente (96).
Il down-sampling dei dati di sequenze genomiche disponibili da località sovrarappresentate può essere utile per indagare se le conclusioni possono essere relativamente robuste in questo senso (97).
L’uso delle statistiche di supporto del fattore di Bayes aggiustato (98) può fornire un ulteriore aiuto nel determinare se gli eventi di transizione sono supportati a causa di un campionamento geograficamente distorto.
-Campionare solo alcune aree di un focolaio può risultare in ricostruzioni inaccurate della storia della dispersione e delle stime della velocità di dispersione all’interno del quadro filogeografico continuo.
Attualmente si stanno valutando modi per ridurre l’impatto del campionamento distorto (99).
-Le informazioni sulla localizzazione del paziente sono spesso limitate alla subunità amministrativa, per esempio, comune.
È spesso opportuno considerare l’incertezza associata a un campionamento quando si usa l’approccio filogeografico continuo.
Per esempio, piuttosto che utilizzare le coordinate geografiche della città più vicina, l’intero poligono corrispondente comune può essere usato per definire un’area dalla quale le coordinate per quel campione possono essere selezionate casualmente.
Anche ripetere questo campionamento casuale durante l’analisi aiuta (100, 101).
5.4.4 Valutazione della trasmissione
I metodi descritti nella sezione precedente possono anche essere utilizzati per indagare i fattori che hanno guidato la dispersione dei virus (97).
Nei modelli filogeografici discreti (compresi quelli implementati come modelli strutturati di coalescenza e modelli multi-tipo di nascita-morte), le informazioni su coppie di aree definite è usata come predittore del tasso di migrazione del lignaggio del virus tra quelle aree.
Le informazioni potrebbero includere la mobilità umana, le caratteristiche della popolazione, come la densità, e la vicinanza geografica.
Gli eventi di dispersione dedotti dalla ricostruzione filogeografica continua ricostruzione filogeografica continua possono anche essere analizzati per determinare se sono influenzati dal “paesaggio” di fattori ambientali o umani attraverso i quali si verificano.
Al momento in cui scriviamo, tali analisi non sono ancora state applicate alla SARS-CoV-2.
Identificare i di trasmissione può aiutare a definire nuove strategie per prevenire la diffusione.
Per esempio, per il virus Ebola questo metodo è stato usato per stabilire che il virus aveva più probabilità di diffondersi tra paesi che condividono i confini terrestri (102) e questo metodo è stato successivamente utilizzato per valutare l’effetto delle effetto delle misure adottate (103).
Limitazioni. Questi approcci sono computazionalmente impegnativi, coinvolgono grandi set di dati di migliaia di genomi e richiedono giorni o settimane per essere completati.
L’uso di una distribuzione pre-stimata di alberi empirici può ridurre il tempo computazionale richiesto ed è particolarmente appropriato per l’esplorazione preliminare dei dati.
Anche il sottocampionamento di cladi specifiche o il sottocampionamento casuale possono ridurre il carico computazionale (sezione 6.8.1).
C’è anche un limite computazionale al numero di aree definite che possono essere incluse nel modello.
Alcuni modelli hanno una flessibilità relativamente limitata nell’identificare i fattori che possono guidare la trasmissione della SARS-CoV-2 in tempi e luoghi diversi.
I modelli epocali implementati all’interno del quadro filogeografico discreto (104) possono essere appropriati per indagare gli effetti variabili nel tempo di diversi fattori in cui possono essere predefiniti periodi di tempo significativi.
Tuttavia, le misure di controllo cambiano rapidamente in molti paesi in tempi diversi.
Questo potrebbe limitare la capacità di definire epoche epidemiologicamente utili quando si applicano queste tecniche sopra la scala nazionale o regionale.
È necessaria una sostanziale competenza per specificare questi modelli in modo appropriato e per interpretare le stime risultanti.
È probabile che le tecniche per valutare gli effetti degli interventi siano applicate retrospettivamente, forse mesi dopo l’intervento.
Le analisi dell’effetto degli interventi che hanno avuto successo nel ridurre i casi potrebbe aiutare a guidare le strategie future nei paesi in cui l’epidemia sta in corso.
Il campionamento distorto può influenzare i risultati (sezione 5.4.3).
5.4.5 Discernere il coinvolgimento di altre specie
Un certo numero di specie animali non umane possono infettarsi naturalmente con la SARS-CoV-2, inclusi gatti, cani e visoni (105-107).
Quando si osservano coppie epidemiologicamente collegate di un umano e di un animale infetto, non è possibile determinare la direzionalità dell’infezione tra di loro.
Quando più animali sono infettati, le indagini filogenetiche di clustering possono essere utilizzate per dimostrare che gli animali si sono infettati attraverso vie diverse, come è stato fatto per i visoni in due allevamenti nei Paesi Bassi (106).
Un forte supporto (alto supporto bootstrap o supporto posteriore) per la collocazione di una sequenza del genoma della SARS-CoV-2 prelevata da un uomo all’interno di un cluster di sequenze multiple campionate dai visoni sarebbe coerente con l’uomo che viene infettato dagli animali.
Se l’ordine dei rami non è fortemente supportato, la direzionalità non può essere dedotta in modo robusto.
Metodologie più estese che impiegano la ricostruzione formale di tratti ancestrali discreti discreta potrebbero anche essere eseguite (sezione 5.4.3).
5.4.6 Discernere le catene di trasmissione tra pazienti usando la diversità virale intra-ospite
Come menzionato in precedenza, poiché le sostituzioni nucleotidiche sembrano verificarsi circa ogni 2 settimane per la SARS-CoV-2, rispondere alle domande epidemiologiche su una scala temporale più fine sarà impegnativo.
Per altri virus, la variazione genetica intra-ospite tra i virioni è stata usata per aumentare la risoluzione alla quale la trasmissione può essere dedotta filogeneticamente.
Virus intra-ospite varianti minoritarie (varianti che si verificano a bassa frequenza all’interno di un individuo) che vengono trasmesse
tra i pazienti forniscono informazioni che sono oscurate con il genoma di consenso.
L’analisi di queste varianti è stata usata per migliorare la comprensione delle vie di trasmissione di molti diversi virus (108, 109).
La variazione intra-ospite esiste per i coronavirus che sono strettamente correlati alla SARS-CoV-2, come MERS-CoV (110).
Mentre i dati attuali (limitati) supportano l’esistenza di variazioni genetiche intra-ospite nella SARS-CoV-2, ad oggi ci sono pochissime serie di dati sulla variazione all’interno dell’ospite da cluster epidemiologici noti che potrebbero essere utilizzati per determinare se questa variazione è trasmessa tra i pazienti (111).
Se non lo fosse, l’uso di queste tecniche non sarebbe possibile.
Sono necessarie analisi bioinformatiche e filogenetiche specialistiche per analizzare il virus intra-ospite variazione del virus intra-ospite.
Data l’attuale mancanza di comprensione dell’entità della variazione intraospite della SARS-CoV-2 o della sua trasmissibilità, queste analisi specialistiche non sono trattate qui.
Limitazioni. Molti set di dati di sequenze genomiche del virus non saranno appropriati per queste analisi.
Il sequenziamento Sanger o il sequenziamento di prossima generazione che utilizza dispositivi che hanno alti tassi di errore di sequenziamento per lettura tassi di errore di sequenziamento senza replicazione (112), non forniranno informazioni sufficienti sulle variazioni intra-ospite.
Il rumore causato dalla contaminazione incrociata dei campioni e dagli errori di sequenziamento può oscurare i veri segnali.
5.5 Inferenza dei parametri epidemiologici
Il numero di riproduzione, R0, può essere stimato utilizzando modelli genetici di popolazione, come modelli di coalescenza, di coalescenza strutturata e di campionamento nascita-morte.
Questi approcci filodinamici sono tutti basati sul concetto che i parametri epidemici, come R0, influenzano la forma delle filogenesi risolte nel tempo.
I vari approcci si basano su presupposti diversi, hanno requisiti di dati leggermente diversi e sono suscettibili di diverse forme di distorsione.
Sono anche appropriati in punti diversi dell’epidemia, a seconda dell’estensione della diffusione geografica e della popolazione studiata.
Nelle primissime fasi della pandemia di SARS-CoV-2, la struttura geografica della popolazione poteva essere largamente ignorata ; le stime di R0 si basavano su dati di sequenza campionati a livello globale con l’approssimazione l’approssimazione che tutti i casi fossero distanti solo poche generazioni dall’epidemia originale di Hubei, Cina (113).
In queste condizioni di campionamento, i modelli di nascita-morte (114) e di coalescenza che si basano su una singola popolazione panmica (mescolanza casuale) possono essere applicati.
Con la dispersione globale della SARS-CoV-2, è diventato possibile e opportuno stimare R0 in diversi paesi, regioni e città.
Una volta che una sostanziale strutturazione geografica delle cladi indicativa della predominanza della trasmissione intraregionale era filogeneticamente evidente (sezione 5.4.3), i modelli di campionamento nascita-morte e coalescenza basati su una popolazione panmatica sono diventati non validi.
I metodi sono stati quindi applicati a livello di singoli cluster filogenetici identificati che rappresentano un lignaggio che circola nella comunità.
Questo richiede una definizione a priori dei cluster filogenetici cladi.
È possibile utilizzare modelli genetici di popolazione più complessi che tengono conto di più importazioni multiple di lignaggi di SARS-CoV-2 e la trasmissione comunitaria ; tali modelli non richiedono una definizione a priori dei cluster.
Queste analisi sono possibili usando modelli di coalescenza strutturati o modelli di nascita-morte di tipo multiplo (85). tipo di modelli di nascita-morte (85, 113), che potenzialmente fanno uso di più metadati clinici e demografici metadati clinici e demografici che influenzano i tassi di trasmissione o i modelli di trasmissione.
Il loro sviluppo e implementazione richiedono una notevole competenza e una buona conoscenza della modellistica epidemiologica.
I requisiti computazionali sono molto più alti che per molte altre applicazioni filogenetiche o filodinamiche.
Limitazioni. I professionisti dovrebbero essere consapevoli della robustezza dei diversi metodi in relazione a diverse forme di distorsione.
Tutti i metodi sono fallibili in presenza di un campionamento distorto, come si verifica quando si sequenzia da catene di trasmissione identificate attraverso il tracciamento dei contatti o piccoli cluster identificati epidemiologicamente.
L’errata specificazione del modello è una fonte di distorsione per tutti i metodi.
Questo viene migliorato con metodi coalescenti strutturati più complessi, ma questi richiedono uno sforzo di calcolo maggiore.
I singoli metodi sono influenzati in modo diverso dalle diverse fonti potenziali di distorsione.
-I modelli di coalescenza basati su relazioni deterministiche tra R0 e il modello demografico possono fornire una stima distorta di R0 quando la dimensione dell’epidemia è piccola o R0 è vicino a 1 (115) e predominano gli effetti stocastici.
-I modelli di campionamento nascita-morte richiedono un’appropriata parametrizzazione della variazione del tasso di campionamento nel tempo (116). Dato che molti paesi sono stati attivamente testati per la SARS- CoV-2 da prima dell’inizio delle loro epidemie, può essere ragionevole assumere che la proporzione di campionamento sia maggiore di zero per l’intera durata dell’analisi.
Tuttavia, se le strategie di test sono cambiate ad un certo punto durante questo periodo, la proporzione di campionamento dovrà variare in modo simile.
I modelli a coalescenza possono fornire stime più precise rispetto ai modelli di campionamento nascita-morte se il tasso di campionamento varia nel tempo.
-Le analisi basate su cluster identificati a priori non possono essere considerate rappresentative della comunità nel suo complesso perché trascurano le piccole catene di trasmissione che non vengono campionate o che sono al di sotto della soglia di dimensione richiesta per l’analisi.
Quindi, i cluster osservati sono quelli che sono cresciuti con più successo.
I numeri di riproduzione dei cluster sono probabilmente più grandi in questi cladi che nella comunità nel suo complesso.
-Quando si imposta un’analisi che presuppone l’assenza di una popolazione strutturata, è fondamentale assicurarsi che ci sia un solo parametro R0 nel periodo di tempo coperto dall’albero che mette in relazione i campioni. Se la quarantena o altre misure sono state introdotte durante il periodo di studio, sarà necessario escludere le sequenze raccolte dopo l’istituzione di queste misure, oppure includere tutte le sequenze ma permettere che il parametro R0 cambi nel tempo.
Molti approcci, compresi i modelli di nascita-morte che sono implementati nel pacchetto software Birth Death Skyline Model (BDSKY) (114), richiedono l’incorporazione esplicita di informazioni informazioni precedenti per fissare alcuni parametri a valori noti e, quindi, per migliorare la trattabilità computazionale.
Tipicamente, è comune fissare un parametro che può essere verificato dai dati clinici, come il tasso al quale gli individui infetti diventano non infettivi. Il parametro precedente dovrebbe essere condotto con attenzione per evitare potenziali fonti di distorsione.
Condurre analisi utilizzando specifiche di priorità alternative può aiutare a determinare quanto sensibili i risultati filodinamici sono sensibili al parametro prioritario specificato.
5.5.2 Scala del focolaio nel tempo e rapporto infezione/caso segnalato
Nella genetica di popolazione tradizionale, la dimensione effettiva della popolazione (il numero di individui in una popolazione che contribuiscono con successo alla progenie nella generazione successiva) è stimata piuttosto che la dimensione assoluta della popolazione del virus (numero totale di virioni) o il numero di individui infetti (dimensione dell’epidemia).
La dimensione effettiva della popolazione può essere usata per identificare i cambiamenti relativi nella dimensione dell’epidemia nel tempo se sono soddisfatte certe condizioni.
La stima della dimensione assoluta dell’epidemia dai dati genetici è stata tentata solo recentemente ed è un’area attiva di sviluppo metodologico filodinamico sviluppo metodologico. Una varietà di metodi sperimentali è stata applicata nell’attuale epidemia di COVID-19.
In generale, qualsiasi metodo per ricostruire le dimensioni dell’epidemia dovrebbe tenere conto dei principali fattori che influenzano la diversità genetica all’interno del quadro di campionamento, tra cui : struttura geografica struttura geografica, varianza nei tassi di trasmissione, crescita esponenziale e dinamica non lineare della popolazione, e la distribuzione del tempo di generazione (117).
Tre diversi approcci e le loro limitazioni sono evidenziati di seguito.
– In alcune situazioni, la dimensione effettiva della popolazione stimata con modelli coalescenti può essere tradotta in dimensioni epidemiche.
Per esempio, Koelle & Rasmussen hanno derivato una formula per fare così che fa uso di stime indipendenti di R0 e della varianza nei tassi di trasmissione sotto equilibrio epidemico (118).
Questo è stato successivamente esteso a uno scenario con crescita esponenziale crescita esponenziale da Li, Grassly e Fraser (117).
Limitazioni. Quest’ultimo approccio è limitato al primo periodo epidemico con crescita esponenziale ed entrambi gli approcci possono essere inappropriati quando c’è una sostanziale strutturazione geografica o demografica nella trasmissione del virus.
Per esempio, quando la trasmissione del virus avviene separatamente in due luoghi diversi senza una trasmissione sostanziale tra i luoghi, potrebbero essere necessari due diversi valori di R0.
– In un quadro di nascita-morte come il BDSKY, la proporzione di campionamento può essere dedotta, e può essere combinata con il numero di sequenze per ottenere una stima grezza del numero cumulativo di casi.
Limitazioni. Anche se forse è un mezzo utile per ottenere una stima rapida, questo approccio è limitato, in particolare per campioni di piccole dimensioni, poiché ignora l’effetto della stocasticità nella procedura di campionamento.
È applicabile a una popolazione non strutturata/panmica, come in un singolo cluster filogenetico o all’inizio dell’epidemia.
Questi approcci non tengono conto di un’alta varianza nei tassi di trasmissione.
Esistono approcci meno limitati, come l’uso di particelle per campionare la curva di prevalenza assoluta direttamente come parte dell’inferenza nascita-morte (119).
– I modelli di coalescenza strutturati che sono implementati nel pacchetto PhyDyn (120) per il software di filogenetica BEAST2 sono stati sviluppati per stimare la dimensione dell’epidemia tenendo conto di variabili come la struttura geografica, le dinamiche non lineari e l’alta varianza nei tassi di trasmissione.
Limitazioni. Questi metodi richiedono competenze di modellazione epidemiologica e hanno alti requisiti computazionali.
Fattori come la selezione naturale, la struttura geografica non modellata struttura geografica, o ricombinazione genomica possono ancora confondere le stime.
6. Guida pratica sugli aspetti tecnici del sequenziamento genomico e analisi della SARS-CoV-2
Le considerazioni generali per l’implementazione di un programma di sequenziamento sono state discusse nella sezione 3.
Questa sezione si concentra sui diversi aspetti tecnici dei progetti di sequenziamento genomico per COVID-19.
6.1 Strategie di campionamento del genoma e disegno dello studio
Le strategie di campionamento del genoma dipendono dalle risposte che si cercano. (N.d.T. : lo avevamo capito !)
Per esempio, l indagine sulla trasmissione nosocomiale o la valutazione dei risultati della ricerca dei contatti (sezione 5.4.1) può richiedere un ampio campionamento genomico della maggior parte dei pazienti identificati nel cluster epidemiologico di interesse, così come campioni che non fanno parte del cluster in esame.
I campioni dall’esterno del cluster sono importanti per sostenere l’ipotesi che i campioni del cluster siano epidemiologicamente legati più strettamente l’uno all’altro che ad altre infezioni comunitarie.
Al contrario, gli approcci filodinamici (sezioni 5.4.2-5.5 e Tabella 1) sono facilmente influenzati da un campionamento non campionamento non casuale di tutti i casi confermati ma, tipicamente, tollerano un campionamento relativamente scarso di una bassa proporzione di tutti i casi.
In particolare, i modelli filodinamici assumono che le sequenze siano raccolte uniformemente a caso da ogni compartimento nel modello sottostante. Questo presupposto può essere facilmente violato se, per esempio, i campioni sono raccolti come risultato della ricerca per contatto.
Per gli approcci filodinamici, i genomi dei virus dovrebbero quindi idealmente essere sequenziati in proporzionalmente all’incidenza reale dei casi. Come questo possa essere meglio approssimato nella pratica può variare.
Dove la copertura diagnostica è buona in un’intera regione, si potrebbe sequenziare un sottoinsieme casuale di campioni diagnostici positivi e residui. Tuttavia, in molti contesti le diagnosi cliniche sono condotte in modo non casuale, anche quando per identificare i casi si ricorre a un’ampia ricerca di contatti.
La proporzione di casi da cui sono disponibili campioni clinici può cambiare nel tempo man mano che vengono implementati diversi regimi di campionamento.
In alcuni paesi, i campioni positivi non rifletteranno la reale distribuzione delle infezioni a causa delle disparità nelle risorse o nell’accessibilità tra i luoghi (ad esempio, un numero sproporzionatamente inferiore di campioni dalle aree rurali a causa delle difficoltà nel trasporto dei campioni per i test centralizzati).
In questi paesi, può essere più appropriato selezionare deliberatamente un set di campioni per il sequenziamento che compensi le distorsioni note nel campionamento.
Per esempio, se la segnalazione di casi sospetti è nota per essere più rappresentativa della segnalazione di casi confermati, potrebbe essere appropriato selezionare campioni da tempi e luoghi diversi in proporzione al numero di casi sospetti piuttosto che al numero di casi confermati.
Non è possibile dare raccomandazioni universalmente appropriate per il sequenziamento della SARS-CoV-2, poiché le decisioni dipenderanno dal contesto del focolaio e dalle domande a cui si vuole rispondere.
I requisiti chiave di requisiti sono elencati nella Tabella 1.
Inoltre, l’allegato 1 evidenzia i tipi di strategie di campionamento strategie di campionamento che sono state utilizzate in altri focolai virali per le specifiche applicazioni filodinamiche considerate nel Box 1.
Tuttavia, il numero di campioni richiesti per la SARS-CoV-2 sarà diverso da quello presentati a causa delle differenze nella diversità virale di base, nella lunghezza del genoma, nel tasso di sostituzione e dinamiche di trasmissione.
6.2 Metadati appropriati
Per garantire che i dati genomici della SARS-CoV-2 siano il più utili possibile, dovrebbero essere accompagnati da metadati appropriati.
Curare i metadati e condividerli localmente o pubblicamente può richiedere tempo. ma entrambi sono parte integrante di qualsiasi pipeline di sequenziamento. Le risorse necessarie dovrebbero essere assegnate quando lo studio viene progettato.
I metadati dovrebbero includere come minimo assoluto la data e il luogo di raccolta del campione.
Tuttavia, il rilascio di metadati aggiuntivi aumenta notevolmente le potenziali applicazioni di una sequenza genomica sequenza genomica.
Dove possibile, quindi, le informazioni sul tipo di campione e su come la sequenza è stata ottenuta in laboratorio dovrebbe essere inclusa (Tabella 2).
Campioni duplicati dallo stesso individuo o sequenze duplicate dallo stesso campione dovrebbero essere chiaramente identificati.
Informazioni demografiche e cliniche, come età, sesso, presenza di comorbilità, gravità della malattia la gravità e l’esito della malattia, e i collegamenti ad altre sequenze nel database, sono incoraggiati quando tali informazioni non rischiano di identificare il paziente.
Un consenso globale su formati specifici per i metadati (come la data) permetterebbe ai dati di sequenze genomiche di sequenze genomiche provenienti da molti laboratori diversi di essere rapidamente compilati in set di dati più grandi e ridurre l’ambiguità.
Alcuni depositi di genoma di consenso, tra cui GISAID, già pongono restrizioni di formato su alcuni campi.
Se i depositi di dati non impongono già dei formati, le restrizioni di formato per SARS-CoV-2 mostrate nella Tabella 2 sono suggerite. La Tabella 2 evidenzia anche esempi di analisi che richiedono la fornitura di metadati specifici.
L’OMS incoraggia fortemente la rapida condivisione pubblica di sequenze e metadati (sezione 4). Tuttavia, è vitale proteggere l’anonimato del paziente. I laboratori dovrebbero considerare attentamente se i pazienti potrebbero essere identificati se tutti i metadati disponibili sono condivisi insieme. Dove sono stati osservati pochi casi di COVID-19 sono stati osservati, c’è un rischio maggiore che l’anonimato del paziente sia compromesso e quindi in genere si possono condividere meno dati. Laddove sia giudicato inappropriato condividere metadati dettagliati metadati dettagliati tramite repository disponibili al pubblico, può comunque essere opportuno concedere l’accesso a
un piccolo numero di utenti attraverso piattaforme sicure sviluppate localmente.
Quando non è possibile condividere tutti i metadati senza rischiare la riservatezza del paziente, i dati che sono più utili per gli studi globali dovrebbero essere condivisi di preferenza. Per esempio, il campionamento luogo, data e storia del viaggio sono più utili per gli studi filodinamici rispetto all’età o al sesso del paziente
(Tabella 2).
Alcuni laboratori scelgono di aggiungere dei jitter (rumore) alle date fornite per diminuire la possibilità che che i pazienti possano essere identificati. Questo può essere ottenuto con una serie di metodi, per esempio scegliendo una data falsa entro 5 giorni dalla data di raccolta del campione o utilizzando la data di sequenziamento come data del campione.
Tali pratiche influenzano negativamente l’inferenza filogenetica basata sull’orologio molecolare filogenetico basato sull’orologio molecolare e dovrebbero idealmente essere evitate.
Se, tuttavia, questa pratica viene seguita, le informazioni su come esattamente è stata selezionata la nuova data dovrebbero essere fornite come nota.
6.3 Considerazioni logistiche
6.3.1 Ubicazione
La decisione su dove basare un laboratorio di sequenziamento dovrebbe essere attentamente considerata.
Il sequenziamento dovrebbe essere generalmente condotto da istituzioni con la necessaria esperienza e infrastrutture per il sequenziamento di prossima generazione.
Se tale infrastruttura non è disponibile, la decisione di dove ospitare il laboratorio di sequenziamento dovrebbe prendere in considerazione l’impatto su altri lavori svolto dal laboratorio.
Per esempio, integrare il sequenziamento in un laboratorio diagnostico esistente laboratorio diagnostico esistente può consentire un tempo di risposta più breve, ma questo guadagno potenziale dovrebbe essere bilanciato contro il rischio di interrompere altre operazioni nel laboratorio, che potrebbe già essere in procinto di aumentare la sua capacità diagnostica per la SARS-CoV-2.
Dovrebbe essere data anche un’attenta considerazione alla disponibilità di spazio e di attrezzature.
Quando la manipolazione degli ampliconi PCR è necessaria per il sequenziamento (ad esempio i metodi descritti nella sezione 6.5.4), è importante ridurre il potenziale di contaminazione degli ampliconi attraverso un’appropriata gestione del laboratorio.
La separazione fisica delle aree che saranno utilizzate per la manipolazione pre e post manipolazione post-PCR del materiale SARS-CoV-2, e un flusso a senso unico di personale e materiali dalle aree pre- e post-PCR, sono fortemente consigliati.
Dove non sono già disponibili aree separate disponibili, i laboratori potrebbero adottare strategie, come l’acquisto e l’uso di glovebox separati o per le attività pre o post-PCR.
L’attrezzatura dovrebbe essere idealmente designata per l’uso solo con materiale pre- o post-PCR, e i reagenti necessari dovrebbero idealmente essere conservati separatamente (per esempio in diversi congelatori o diversi laboratori) per ridurre il rischio di contaminazione.
Come per tutti i sequenziamenti, i controlli negativi sono preziosi per rilevare la contaminazione.
6.3.2 Prevenzione e biosicurezza
Le valutazioni del rischio dovrebbero sempre essere condotte per valutare la biosicurezza e la biosicurezza.
I risultati di tali valutazioni del rischio dovrebbero essere comunicati ai lavoratori coinvolti nei processi pertinenti.
I singoli laboratori dovrebbero sempre condurre valutazioni di rischio locali per ogni fase del loro protocollo SARS-CoV-2.
La legislazione internazionale, nazionale e locale dovrebbe essere consultata per garantire la manipolazione sicura del materiale SARS-CoV-2.
L’OMS ha emanato ampie linee guida sulla biosicurezza (121).
I campioni devono essere inattivati il prima possibile (di solito prima dell’estrazione dell’RNA) usando metodi chimici che preservino la qualità dell’RNA.
I metodi usati per estrarre l’RNA prima di NAATs diagnostici sono generalmente appropriati per il sequenziamento.
Come per la maggior parte delle NAAT, il calore inattivazione prima dell’estrazione del campione non è raccomandata perché rischia di danneggiare l’integrità dell’RNA integrità.
6.3.3 Considerazioni etiche
Le revisioni etiche dovrebbero essere condotte per assicurare che i pazienti abbiano dato il consenso appropriato per la raccolta e il sequenziamento dei campioni, e per considerare il successivo uso, la conservazione e la pubblicazione dei dati.
Alcuni approcci di sequenziamento, come la metagenomica, genereranno dati genomici umani.
Qualsiasi sequenza genomica umana dovrebbe essere rimossa dal set di dati virali attraverso una pipeline di analisi automatica automatica il più presto possibile, senza operazioni manuali da parte del personale (vedi sezione 6.7.1), a meno che non siano stati ottenuti l’approvazione etica e il consenso esplicito del paziente al trattamento dei dati genetici umani. ottenuto.
Se i dati personali o umani devono essere conservati, si raccomanda vivamente un’adeguata crittografia di tutti questi file. altamente raccomandata.
Le revisioni etiche dovrebbero determinare il massimo possibile di metadati rilevanti che possono essere condivisi senza rischiare la riservatezza del paziente.
6.3.4 Risorse umane
È importante assicurare che ci sia personale sufficiente per sostenere tutti gli aspetti del programma di sequenziamento programma di sequenziamento, dal campionamento clinico alla comunicazione dei risultati e alla condivisione di sequenze e metadati.
Il costo di un programma di sequenziamento dovrebbe includere i costi del personale, così come i costi dei dispositivi di protezione personale, dei materiali di consumo, dell’acquisto e della manutenzione di altre attrezzature e dell’architettura computazionale.
Se diversi laboratori o istituti sono coinvolti in indagini in collaborazione, può essere utile ottenere un accordo scritto sulle responsabilità di ciascun laboratorio (per esempio in relazione al finanziamento, al personale che può essere impegnato e al lavoro da eseguito) e i benefici attesi prima che il progetto abbia inizio.
Il contenuto di tali accordi varierà; gli accordi di collaborazione istituzionale esistenti o gli accordi di trasferimento di materiale possono fornire modelli appropriati.
Le implicazioni in termini di risorse umane di qualsiasi programma di sequenziamento pianificato dovrebbero essere considerate con riferimento ai modelli di lavoro previsti.
In generale, un modello di lavoro normale dovrebbe essere incoraggiato in modo da evitare il burnout del personale.
La probabilità di malattia e indisponibilità del personale nel nel contesto della pandemia COVID-19 dovrebbe anche essere presa in considerazione.
I tentativi di costruire capacità extra capacità extra nel flusso di lavoro dovrebbero essere considerati in anticipo, pur riconoscendo che la generazione di genomi di patogeni da campioni clinici richiede un team multidisciplinare con set di competenze specifiche.
L’intensità e la prevedibilità del carico di lavoro dipendono dagli obiettivi del progetto (Tabella 3).
I laboratori diagnostici sono spesso centrali per l’identificazione dei casi positivi e per la sicurezza trattamento e conservazione dei campioni dei pazienti.
Se è previsto un progetto di sequenziamento su larga scala, è raccomandato che un rappresentante del laboratorio diagnostico sia designato per fare da tramite direttamente con il team di sequenziamento per assicurare un recupero efficiente dei campioni e dei metadati rilevanti per applicazioni a valle.
6.4. Scelta del materiale appropriato per il sequenziamento
6.4.1 Materiale per il sequenziamento
L’acquisizione di una quantità sufficiente di RNA di SARS-CoV-2 di alta qualità aiuta a massimizzare la resa del sequenziamento e la qualità finale dei dati di sequenza del genoma.
La quantità e la qualità di un campione di RNA sono influenzate da: scelta del campione clinico ; trattamento del campione clinico; metodo di isolamento dell’RNA virale RNA virale; e la competenza tecnica del personale.
Quando sono disponibili diversi tipi di campioni, è vantaggioso selezionarne uno che abbia un’elevata carica virale e bassi livelli di contaminanti di materiale genetico umano o batterico (Tabella 4).
Tali campioni possono essere sequenziati utilizzando sia tecniche metagenomiche che tecniche mirate alla SARS-CoV-2 (sezione 6.5).
Alcuni materiali, come le feci, possono richiedere la centrifugazione e la filtrazione prima di l’estrazione dell’RNA virale, per eliminare il materiale cellulare umano o batterico che può ridurre la sensibilità del sequenziamento.
In molti contesti, gli unici campioni ordinariamente disponibili per il sequenziamento del genoma del virus saranno campioni diagnostici residui.
I campioni raccolti per la diagnostica NAAT sono in genere anche appropriati per il sequenziamento (77).
I tamponi nasali, i tamponi della gola e la saliva sono risultati avere elevati carichi virali poco dopo la comparsa dei sintomi e fino a 25 giorni dopo (140, 158, 159).
La carica virale della SARS-CoV-2 e l’abbondanza di RNA virale nei campioni è normalmente più alta nella prima settimana dopo la comparsa della malattia (158, 160).
Se possibile, gli isolati per il sequenziamento dovrebbero essere selezionati da campioni positivi che sono già stati trattati da un laboratorio di diagnostica molecolare (Fig. 3).
Condividere le risorse in questo modo evita la duplicazione del lavoro nel trattamento dei campioni e nell’estrazione dell’acido nucleico e può quindi far risparmiare risorse umane e di altro tipo e costi.
Alcuni kit commerciali di diagnostica molecolare usano lisati virali come input, e non permettono di conservare l’RNA estratto.
In tali casi in cui i componenti del buffer di lisi commerciale non sono divulgati, può essere estremamente impegnativo riutilizzare i lisati preparati con altri kit di estrazione commerciale e può essere necessario eseguire l’inattivazione fresca e l’estrazione direttamente dal campione clinico originale.
La divulgazione dei componenti dei buffer di lisi commerciali aiuterebbe i ricercatori a sviluppare strategie per riutilizzare lisati già inattivati per l’uso in successive attività di sequenziamento.
Un sistema pratico ed efficace di identificazione del campione dovrebbe essere usato se i campioni si spostano tra i laboratori; idealmente, la stessa identificazione del campione dovrebbe essere usata in tutti i laboratori.
Fig. 3. Esempio di flusso di lavoro per il recupero dei campioni da un laboratorio diagnostico.
Conservare l’RNA virale è importante per la produzione di dati di sequenza di alta qualità.
Questo può essere ottenuto mantenendo una catena del freddo tra la raccolta del campione e l’analisi, riducendo il numero di volte che l’RNA o i campioni sono congelati e scongelati, e riducendo al minimo il tempo tra la raccolta del campione e il sequenziamento.
RNA che viene conservato o spedito a 4 °C per più di pochi giorni è improbabile che sia di qualità sufficientemente elevata per il sequenziamento a meno che non è stato prima conservato in una soluzione di stabilizzazione RNA.
La qualità sarà sostanzialmente più alta se l’RNA può essere conservato a – 20 °C o preferibilmente a -80 °C.
Lisati virali in genere non possono essere conservati a 4 °C per tanto tempo come estratto RNA.
Molti protocolli di sequenziamento includono passaggi che migliorano la capacità di conservazione di un campione, compresa la trascrizione inversa dell’RNA in cDNA, o la sintesi del secondo filamento/generazione di amplicati PCR a doppio filamento di DNA.
DNA a doppio filamento di ampliconi PCR. Gli ampliconi PCR possono essere conservati a 4 °C per molti mesi senza riduzione della qualità del sequenziamento.
In alcuni contesti, può quindi essere opportuno eseguire questi passaggi rapidamente dopo le PCR diagnostiche, in modo che il materiale possa essere conservato o spedito con meno vincoli di temperatura prima della preparazione della libreria.
6.4.2 Campioni di controllo
I campioni di controllo negativo, come il buffer o l’acqua, dovrebbero sempre essere inclusi in ogni sequenziamento che contiene più campioni.
Dovrebbero essere inclusi nella prima fase possibile e dovrebbero procedere con i campioni attraverso tutte le fasi della pipeline di sequenziamento. Questo è estremamente importante per escludere la contaminazione durante una corsa di sequenziamento che si verifica in laboratorio o durante l’elaborazione bioinformatica.
I campioni di controllo positivo con sequenze genetiche note possono essere utili per convalidare le pipeline bioinformatiche appena adottate o adattato pipeline bioinformatiche per la chiamata del consenso, ma non è necessario che siano inclusi in ogni sequenziamento.
6.5 Arricchimento del materiale genetico SARS-CoV-2 prima della preparazione delle librerie
Le strategie di sequenziamento per la SARS-CoV-2 includono approcci metagenomici, che non richiedono conoscenza preliminare della sequenza genomica, e approcci mirati, che si basano sulla conoscenza del del genoma.
Entrambi gli approcci tentano tipicamente di arricchire il materiale genetico della SARS-CoV-2 in relazione ad altri RNA/DNA prima del sequenziamento.
Se è disponibile una quantità sufficiente di RNA residuo ed è stato conservato in modo appropriato (sezione 6.4.1), la maggior parte degli approcci può essere eseguita utilizzando l’RNA estratto per saggi diagnostici.
Sono già stati condivisi molti protocolli diversi per il sequenziamento di SARS-CoV-2 il sequenziamento.
Alcuni di questi sono evidenziati qui di seguito ; altri sono stati raccolti dagli United States Centers for Disease Control and Prevention (CDC) (161) (N.d.T. : ma se hanno di non possederli, dove stanno ?).
6.5.1 Analisi metagenomica di campioni clinici non coltivati
I protocolli metagenomici permettono il sequenziamento non mirato dell’acido nucleico in un campione, compreso il materiale genomico materiale genomico virale se presente (162).
Questi protocolli offrono un approccio senza ipotesi al patogeno, in quanto richiedono poca conoscenza preliminare del patogeno di interesse (163).
La deplezione dell’ospite o di altro materiale genetico non-SARS-CoV-2 in un campione porta ad una maggiore proporzione di letture di SARS-CoV-2 nei dati di sequenza generati e quindi una maggiore possibilità di recuperare un genoma completo.
Gli approcci metagenomici alla SARS-CoV-2 includono quindi tipicamente fasi per rimuovere le cellule ospiti e batteriche, attraverso la centrifugazione o la filtrazione prima dell’estrazione dell’RNA o la rimozione chimica o enzimatica del DNA/RNA indesiderato.
Questo è più facile per i campioni liquidi campioni liquidi, da cui le cellule possono essere più facilmente separate, come il lavaggio broncoalveolare (Tabella 4).
RNA ribosomiale (rRNA) e il contenuto di DNA sono anche comunemente impoverito durante la biblioteca preparazione per il sequenziamento dell’RNA del virus, e RNA vettore è spesso omesso da estrazioni o sostituito con poliacrilammide lineare.
Nonostante tali misure, i campioni possono ancora contenere elevati quantità di DNA/RNA ospite off-target che può anche essere sequenziato. Approcci metagenomici quindi in genere traggono vantaggio dall’inserimento di campioni con un alto carico virale (tale che una ragionevole proporzione del materiale genetico nel campione è virus).
In alternativa, un gran numero di letture di solito deve essere generato ; in questo modo, anche se il materiale genetico della SARS-CoV-2 rappresenta solo una piccola parte delle letture, sarà comunque possibile ottenere l’intero genoma del virus.
Il sequenziamento metagenomico produce tipicamente un alto numero di letture fuori bersaglio, non virali.
È anche spesso (anche se non sempre, a seconda della piattaforma di sequenziamento e del multiplexing) più costoso degli approcci di sequenziamento basati sulla cattura mirata o sull’amplicon, perché devono essere prodotti più dati per generare un genoma di SARS-CoV-2.
Inoltre, le fasi di pretrattamento che sono particolarmente utili per la metagenomica, come la centrifugazione, non vengono in genere eseguite per i test diagnostici molecolari, quindi nuove estrazioni che incorporano fasi di pretrattamento potrebbero dover essere eseguite per il sequenziamento metagenomico.
Gli approcci di sequenziamento mirato (sezioni 6.5.3 e 6.5.4) sono spesso più convenienti e richiedono meno risorse ; possono quindi essere più appropriati quando non sono richiesti i benefici degli approcci metagenomici (ad esempio, scoperta di patogeni, rilevamento di coinfezioni).
Il successo degli approcci metagenomici varia a seconda dei metodi.
Diversi studi hanno dimostrato una rapida riduzione del successo di diverse analisi di sequenziamento metagenomico in campioni con soglie di ciclo (Cts) della PCR in tempo reale (qPCR) superiori a circa 25-30.
Per tali campioni, i metodi PCR multiplex e basati sulla cattura raggiungono copertura costantemente più alta attraverso il genoma rispetto al sequenziamento metagenomico (57, 164).
Il numero di letture di sequenziamento per campione che deve essere generato per ottenere il genoma completo dipenderà dal tipo di campione, dalle procedure di pretrattamento per rimuovere il materiale ospite e dal livello di viremia.
6.5.2 Approcci metagenomici dopo la coltura cellulare
Per i campioni con una bassa carica virale, la proporzione di materiale genetico virale può teoricamente essere aumentata permettendo al virus di replicarsi in coltura cellulare.
Tuttavia, i rischi di biosicurezza associati alla coltura del virus sono significativamente più alti di quelli associati a campioni campioni clinici non coltivati.
Sono richieste strutture di livello 3 di biosicurezza, con procedure aggiuntive estese per garantire una manipolazione e una conservazione sicure. Inoltre, il passaggio in coltura cellulare può portare a mutazioni artificiali mutazioni artificiali nelle sequenze, che non erano presenti nel campione clinico originale.
Questo può avere implicazioni importanti per le analisi successive.
Usare la coltura cellulare al solo scopo di amplificare il materiale genetico del virus per il sequenziamento della SARS-CoV-2 dovrebbe quindi essere evitato, soprattutto ora che sono disponibili altri approcci basati sulla cattura di esche e di ampliconi per migliorare la sensibilità del sequenziamento.
6.5.3 Approcci basati sulla cattura mirata
Dopo la preparazione di una libreria di sequenziamento metagenomico, gli approcci basati sulla cattura che che arricchiscono il materiale genetico della SARS-CoV-2 possono essere eseguiti prima del sequenziamento.
Tali approcci si basano sull’ibridazione del DNA che è stato trascritto inversamente dall’RNA virale, al DNA o all’RNA esche. Queste esche sono progettate per essere complementari alle regioni del genoma della SARS-CoV-2.
Il materiale della libreria off-target che non si è legato con successo a un’esca (ad esempio il DNA dell’ospite) può essere rimosso usando approcci enzimatici o fisici.
Questo riduce la possibilità di rilevare altre co infezioni, ma aumenta il numero previsto di letture di sequenziamento che mappano il genoma del SARS- CoV-2, consentendo a più campioni di essere efficacemente sequenziati insieme in corse multiplex.
Un vantaggio dell’utilizzo di un approccio basato sulla cattura rispetto ad un approccio basato sull’amplificazione PCR (sezione 6.5.4) è che gli approcci basati sulla cattura possono tollerare differenze di sequenza dalla sonda sequenze della sonda del 10-20%.
Questo è superiore al mismatch tollerato dalla PCR, dove una tale divergenza dalle sequenze dei primer comporterebbe un alto rischio di fallimento dell’amplicone.
Gli approcci basati sulla cattura possono quindi essere utilizzati per arricchire con successo le sequenze relativamente divergenti di SARS-CoV-2. Gli approcci basati sulla cattura sono in genere più complessi da stabilire e più costosi degli approcci basati sugli ampliconi PCR.
Diversi pannelli di cattura specifici per la SARS-CoV-2 che sono disponibili in commercio o che possono essere progettati su ordinazione possono portare a un aumento di 100-10.000 volte della sensibilità.
Quando più campioni devono essere sequenziati insieme in un singolo pool, è più conveniente eseguire la cattura su un intero pool di fino a 96 campioni multiplexati dopo il barcoding del campione.
Diversi protocolli pubblicati sono stati convalidati per il sequenziamento di SARS-CoV-2 basato sulla cattura (ad esempio, sulla base di (165)).
6.5.4. Approcci basati su ampliconi mirati
Le PCR che generano ampliconi che coprono l’intero genoma della SARS-CoV-2 possono essere utilizzate per amplificare il materiale virale prima della preparazione della libreria di sequenziamento.
A differenza degli approcci basati sulla cattura, quelli basati sugli ampliconi non tollerano un sostanziale mismatch tra la sequenza mirata e i primer utilizzati.
La diversità genomica mirata deve quindi essere relativamente bassa e/o la sequenza bersaglio sufficientemente nota per consentire ai primer di essere progettati per mirare a regioni genomiche più conservate regioni genomiche più conservate.
Dato che il SARS-CoV-2 è emerso solo recentemente negli esseri umani e quindi mostra una diversità genomica globale relativamente bassa, gli approcci basati sulla PCR sono attualmente molto appropriati per il sequenziamento della SARS-CoV-2.
Tuttavia, il verificarsi di fallimenti degli ampliconi deve essere essere monitorato e i primer devono essere sostituiti laddove il fallimento si verifichi come risultato di sostituzioni nei siti di legame dei primer.
Gli approcci ottimizzati basati sulla PCR sono altamente specifici e sensibili e permettono di ottenere di routine interi genomi del virus SARS-CoV- 2 di essere generati di routine da campioni con valori Ct della PCR fino a 30.
I genomi parziali di genomi parziali possono essere generati di routine da campioni con valori Ct di 30-35.
Tuttavia, questi valori sono un’approssimazione ; il Ct non è un perfetto predittore del successo dell’amplificazione poiché può variare tra diversi metodi diagnostici (166), e l’uso di diversi tipi e qualità di campione influenzerà la sensibilità.
Inoltre, le regioni genomiche prese di mira nei test diagnostici PCR sono in genere molto più corte di quelle utilizzate nei comuni approcci di sequenziamento basati su ampliconi, quindi la degradazione dell’RNA tipicamente influenzerà il sequenziamento basato sulla PCR più della diagnostica PCR.
Dove la diversità genomica diversità genomica è bassa, gli approcci basati sulla PCR sono un modo economico, rapido e conveniente per aumentare la quantità di materiale genetico del virus disponibile in un campione prima del sequenziamento.
Sono stati descritti diversi set di primer per il sequenziamento del genoma completo basato su ampliconi.
Questi ampliconi bersaglio di diverse lunghezze, in genere 400-2000 paia di basi (bp).
Gli ampliconi più lunghi richiedono un minor numero di primer PCR per impalcatura l’intero genoma, ma possono risultare in lacune più grandi nel genoma di consenso nel caso di un fallimento di amplificazione di una coppia di primer.
Gli ampliconi più lunghi sono adatti alle piattaforme long-read, ma richiedono la frammentazione per gli strumenti di sequenziamento short-read.
Lo schema più utilizzato è attualmente l’approccio basato su tiling amplicon progettato dalla rete ARTIC (167).
Mentre il protocollo ARTIC si concentra in gran parte sul sequenziamento nanopore della Oxford Nanopore Technologies, diversi laboratori hanno convalidato l’approccio ARTIC su altre piattaforme di sequenziamento (112, 168).
È fondamentale adottare strategie per prevenire la contaminazione da ampliconi di altri test diagnostici o ulteriori sequenziamenti (sezione 6.3.1).
6.6 Selezione della tecnologia di sequenziamento
Dopo la preparazione iniziale del campione per arricchire il materiale genetico della SARS-CoV-2, le librerie possono essere preparate utilizzando i protocolli di sequenziamento standard che sono appropriati per qualsiasi virus.
Il protocollo dipenderà dallo strumento utilizzato.
Prima di investire nella capacità di sequenziamento per la prima volta, o di adottare una tecnologia alternativa, si dovrebbe considerare il tempo di esecuzione, i costi, la facilità d’uso, la successiva elaborazione dei dati, il rendimento (tasso di produzione dei dati) e la accuratezza delle varie tecnologie (Tabella 5) (vedi anche sezione 6.7).
Il sequenziamento convenzionale (sequenziamento Sanger) può essere usato per sequenziare singoli frammenti (fino a 1000 bp) in reazioni separate.
Il sequenziamento dell’intero genoma della SARS-CoV-2 richiederebbe almeno 30 singoli ampliconi da sequenziare separatamente per ogni campione di paziente.
Il sequenziamento Sanger è quindi probabilmente più utile per il sequenziamento di brevi frammenti di genomi, ad esempio, per riempire i vuoti negli assemblaggi a seguito del sequenziamento di prossima generazione o per studiare la diversità del virus in brevi regioni, come i siti di legame dei primer, a seguito del fallimento di un test diagnostico.
Le piattaforme di sequenziamento di prossima generazione sono più appropriate per il sequenziamento di routine dell’intero genoma.
sequenziamento dell’intero genoma.
Le piattaforme di sequenziamento che sono state comunemente usate fino ad oggi per la SARS-CoV-2 includono quelle di Illumina, IonTorrent e Oxford Nanopore Technologies.
A differenza del sequenziamento Sanger sequenziamento di Sanger, in cui tutte le molecole di DNA in un campione devono avere le stesse sequenze o altamente simili sequenze (per esempio in seguito alla PCR di un singolo amplicone), queste tecnologie permettono il sequenziamento simultaneo sequenziamento di più frammenti del genoma della SARS-CoV-2.
Tutte le piattaforme di sequenziamento di prossima generazione consentono di sequenziare più campioni insieme in una singola corsa.
I vantaggi chiave e le limitazioni di ogni tecnologia sono riassunte nella Tabella 5.
Mentre tutte le piattaforme sono appropriate per generare genomi di consenso di SARS-CoV-2, alcune possono essere più adatte per soddisfare gli obiettivi specifici del programma di sequenziamento.
Per esempio, un tempo di risposta veloce può essere importante per le applicazioni cliniche, mentre l’accuratezza a livello di lettura può essere più importante per lo studio della diversità intra-ospite.
6.7. Protocolli bioinformatici
La selezione di un protocollo bioinformatico appropriato che può elaborare i dati grezzi letti in sequenze di consenso dell’intero genoma è solitamente importante quanto quella della piattaforma di sequenziamento.
L’uso di un protocollo bioinformatico inappropriato potrebbe produrre risultati errati che possono gravemente influire sulle analisi a valle.
6.7.1 Panoramica dei tipici passi bioinformatici
Archiviazione dei dati grezzi letti
Il sequenziamento genera grandi volumi di dati (Tabella 5).
I costi dell’architettura computazionale architettura richiesta per archiviare e gestire questi dati dovrebbero essere considerati quando una pipeline di sequenziamento pipeline di sequenziamento è in fase di sviluppo.
Il volume di dati grezzi prodotti, solitamente memorizzati come file FASTQ (che memorizzano le sequenze genetiche insieme al punteggio di qualità di ogni base nella sequenza), dipenderà dipende dal numero di campioni processati.
I dati a lettura breve che sono stati arricchiti per le sequenze virali sequenze virali, sia tramite cattura dell’esca che tramite amplificazione PCR, possono spesso comprendere 1-2 milioni di letture per campione e richiedere fino a 1 Gb di spazio su disco, a seconda della lunghezza delle letture.
Campioni non arricchiti che sono stati sequenziati metagenomicamente richiederanno tipicamente un numero di letture 100 volte maggiore per ottenere una buona copertura genomica della SARS-CoV-2, poiché la proporzione di letture virali in tali campioni può essere inferiore all’1% delle letture totali (164).
Se la capacità di archiviazione è limitata, l’archiviazione permanente dei dati grezzi potrebbe non essere fattibile.
Mentre è preferibile conservare i dati grezzi letti localmente il più a lungo possibile, non è sempre critico se tale memorizzazione diventa una barriera per ulteriori sequenziamenti.
Un’eccezione è la memorizzazione di dati grezzi da sequenziamento metagenomico o metatranscriptomico, che può contenere informazioni sulla co- infezione con altri virus o batteri.
Tali campioni rappresentano una risorsa preziosa e gli sforzi dovrebbero essere fatti per preservare le informazioni anche se le letture grezze non possono di solito essere conservate in altre circostanze.
Una buona pratica alternativa all’archiviazione locale permanente dei dati di lettura grezzi è quella di caricare i dati in un repository, come SRA (NCBI), DDBJ o ENA.
A meno che la revisione etica non abbia approvato la ricerca e la condivisione di sequenze genomiche umane, e tutti i partecipanti abbiano dato il loro esplicito consenso informato, i dati inviati a depositi pubblici depositi pubblici dovrebbero prima essere privati delle letture di origine umana.
Per gli approcci di sequenziamento mirato alla SARS-CoV-2 SARS-CoV-2, tutte le letture di sequenziamento possono essere mappate sul genoma SARS-CoV-2 e estrarre le letture mappate.
Le letture estratte che successivamente dimostrano di non mappare sul genoma umano genoma umano possono in genere essere inviate agli archivi.
Il software esistente può facilitare questa operazione per diverse piattaforme, per esempio, nanostripper per i dati prodotti usando i dispositivi Oxford Nanopore Technologies (169).
Per i progetti metagenomici in cui uno degli obiettivi è quello di identificare le co-infezioni, le strategie per rimuovere le letture umane sono più complesse.
Alcuni depositi, come SRA, possono rimuovere le letture genetiche umane dai set di dati metagenomici se contattati direttamente.
Le pipeline per rimuovere le letture umane può anche essere stabilito utilizzando software di classificazione tassonomica, come Kraken2 o CLARK (170, 171), o software per la rimozione delle letture che mappano i genomi umani, come GSNAP (172).
I processi per rimuovere le letture umane dovrebbero sempre essere valutati come parte di 48 della revisione etica di qualsiasi progetto e dovrebbero essere ampiamente testati per garantire la loro efficacia.
Altri altri approcci di condivisione dei dati e considerazioni etiche sono trattati più estesamente nella sezione 4.
Assemblaggio del genoma dai dati grezzi
Sono state sviluppate diverse pipeline di software disponibili gratuitamente e ottimizzate per il sequenziamento di SARS-CoV-2.
Molti richiedono una configurazione locale minima e hanno chiare istruzioni per l’uso.
Un utile repository (non esaustivo) di link a pipeline di sequenziamento, compresa la bioinformatica, ove stabilito, è mantenuto dal CDC (161).
Altri pacchetti per il sequenziamento dei virus sono disponibili e sarebbero appropriati dopo un’ampia personalizzazione per SARS-CoV-2.
La pipeline bioinformatica dipenderà dalle fasi di laboratorio precedenti al sequenziamento (ad esempio, l’amplificazione PCR richiede il trimming bioinformatico dei siti dei primer) e dalla piattaforma di sequenziamento e dai reagenti utilizzati.
Le pipeline bioinformatiche spesso includono passaggi simili a quelli mostrati nella Tabella 6.
Tabella 6. Passi comuni nella costruzione del consenso bioinformatico per le due piattaforme di sequenziamento di prossima generazione più comunemente usate.
Indipendentemente dalla pipeline, le varianti nucleotidiche non dovrebbero essere chiamate se il numero di di supporto al sito è inferiore alla profondità richiesta per la fiducia.
Invece, tali siti dovrebbero essere chiamati come basi ambigue (N) nel genoma di consenso finale.
A seconda della precisione delle letture grezze nei metodi scelti, qualsiasi sito con meno di 5-20 letture uniche di supporto non possono essere chiamati accuratamente.
Il livello minimo di contaminazione previsto può essere determinato dal numero di letture di SARS-CoV-2 osservate nel controllo negativo, e i siti dovrebbero essere chiamati solo essere chiamati solo se la profondità supera notevolmente questo livello.
I metodi metagenomici e di cattura sono quantitativi, il che significa che la profondità di lettura dei campioni rifletterà approssimativamente il numero di copie del genoma virale nella libreria di partenza.
Per i campioni con una bassa carica virale, la chiamata delle varianti dovrebbe essere eseguita con cautela, poiché anche un piccolo numero di letture contaminanti può interferire con il segnale del campione.
I controlli negativi dovrebbero essere sequenziati per permettere di valutare la probabilità di contaminazione.
Le varianti nei campioni con Cts elevato che probabilmente hanno pochi numeri di copie di RNA di partenza dovrebbero essere valutate con cautela, perché la presenza stocastica di alcune varianti tra le poche copie presenti può portare a errori artefatti.
Le varianti dovrebbero anche essere considerate con molta cautela se gli enzimi utilizzati durante la trascrizione inversa e/o la PCR inducono frequentemente errori.
Gli enzimi ad alta fedeltà enzimi dovrebbero essere usati dove possibile per proteggersi da tali errori.
6.7.2 Trattare i dati multiplex
È conveniente sequenziare più campioni virali in una singola corsa di sequenziamento.
Questo è generalmente realizzato con l’aggiunta di adattatori unici o codici a barre alle letture di sequenziamento.
Quando i dati grezzi vengono generati, possono essere de-multiplexati assegnando le letture ai campioni con codici a barre corrispondenti.
Il multiplexing introduce una nuova complessità nel processo di controllo della qualità dei risultati bioinformatici, poiché è possibile che i codici a barre siano determinati in modo errato, a causa di un processo noto come index hopping o misassignment dell’indice.
Questi artefatti colpiscono particolarmente campioni con una bassa carica virale, come un piccolo numero di letture contaminanti può avere un effetto sproporzionato sul consenso del genoma.
Per difendersi da questo, si raccomanda che pool multiplexati contengano almeno un controllo negativo (buffer) e, se possibile, un controllo non-SARS- CoV-2, e che il numero di letture non assegnate nella corsa sia determinato sulla base di osservazioni delle letture di controllo nei campioni e nel controllo negativo.
Sistemi unici con doppia indicizzazione (ad es. applicazioni Illumina), o doppia codifica a barre (ad es. applicazioni Oxford Nanopore Technologies e alcune preparazioni Ion Torrent), dovrebbero essere usati dove possibile, e ci dovrebbero essere controlli rigorosi sul demultiplexing dei campioni.
Il demultiplexing dovrebbe essere condotto utilizzando impostazioni rigorose (per esempio, a seconda della tecnologia, richiedendo codici a barre per essere presenti su entrambe le estremità di una lettura di sequenziamento, con pochi o nessun mismatch a quel codice a barre).
6.8 Strumenti di analisi
A metà novembre 2020, 180 000 genomi completi con una buona copertura erano pubblicamente disponibili, e il numero stava aumentando esponenzialmente.
Molti di questi genomi sono probabilmente quasi identici.
Se non è richiesto un genoma completo di migliaia di sequenze quasi identiche, le strategie di down-sampling possono essere impiegate per ridurre le richieste computazionali dell’allineamento e delle analisi successive.
Le strategie di down-sampling devono essere attentamente considerate, in quanto possono influenzare pesantemente le analisi a valle.
Una procedura possibile è quella di eseguire uno strumento di clustering, come cd-hit-est (182), ad un’alta soglia di clustering (> 99% di similarità di sequenza) e costruire un allineamento usando i genomi rappresentativi genomi rappresentativi di questa analisi.
Questo è computazionalmente leggero e verificabile, in quanto viene prodotto un rapporto di clustering viene prodotto un rapporto che indica quali sequenze sono state selezionate per ogni cluster ed elenca l’intera l’appartenenza al cluster.
Un’alternativa può essere quella di selezionare i cladi di interesse da un albero più grande calcolato in precedenza.
Questo può essere una strategia utile, in particolare quando una regione geografica o altre caratteristiche sono di primaria importanza per l’analisi, e l’intera diversità globale dei genomi virali è meno rilevante.
Nextstrain (183) permette di selezionare le cladi da un albero globale e di estrarre successivamente i metadati delle sequenze in quei cladi per essere successivamente estratti e utilizzati per aiutare il sottocampionamento di grandi insiemi di dati disponibili.
Per l’inferenza filogeografica in cui i ricercatori sono interessati a catturare i movimenti del lignaggio del virus movimenti tra i luoghi, ma non all’interno dei luoghi, può essere opportuno eseguire il sottocampionamento basato su criteri filogenetici.
Qui, cladi monofiletici di sequenze dalla stessa posizione potrebbe essere sottocampionata ad una singola sequenza da quel clade, come ulteriori sequenze all’interno del clade non può aggiungere ulteriori informazioni di interesse per quanto riguarda inter-location lineage virale movimenti del lignaggio virale (103, 184).
6.8.2 Allineamenti di sequenze
L’allineamento di migliaia di sequenze del genoma di SARS-CoV-2, molte delle quali includono regioni di ambiguità dovute a genomi parzialmente determinati, è computazionalmente impegnativo.
Pochissimi strumenti esistenti possono far fronte ad allineamenti di questa lunghezza, e vale la pena notare che ogni volta che una nuova sequenza viene generata, essa ha il potenziale di modificare l’allineamento determinato in precedenza.
È possibile utilizzare un software di allineamento, come MAFFT, per aggiungere un piccolo numero di nuove sequenze a un piccolo allineamento esistente con un dispendio computazionale relativamente basso (185).
Allineamenti fino a centinaia di sequenze possono anche essere curati con l’aiuto di esperti, e gli autori di MAFFT (186) offrono questo servizio per gli allineamenti SARS-CoV-2.
Tuttavia, per insiemi di campioni più grandi può essere necessaria una strategia diversa.
La pipeline shiver (187) produce una versione di ogni genoma assemblato che viene allineata per mantenere il posizionamento delle coordinate.
In questo modo, ogni genoma elaborato può essere semplicemente aggiunto in un allineamento crescente senza bisogno di ri-allineare tutte le sequenze ogni volta che viene aggiunta una sequenza, anche se bisogna fare attenzione a non perdere nuove inserzioni.
È spesso opportuno tagliare le regioni non codificanti, comprese le estremità 5′ e 3′ di un allineamento, prima di ulteriori analisi.
Può essere difficile analizzare filogeneticamente tali regioni perché subiscono inserzioni, delezioni e sostituzioni multiple nello stesso sito più frequentemente di regioni codificanti che possono essere sottoposte a una selezione più intensa.
6.8.3 Controllo di qualità
Le sequenze generate dovrebbero sempre essere sottoposte a un controllo di qualità prima di essere utilizzate in qualsiasi analisi.
Le procedure di controllo della qualità dovrebbero essere condotte in diverse fasi, per determinare caratteristiche multiple che possono essere associate a sequenze di scarsa qualità.
La rimozione di sequenze con basi ambigue, indel o frame-shift basati su sequenze non allineate/allineate sequenze non allineate/allineate
La maggior parte degli strumenti software per la costruzione di alberi filogenetici, compresi tutti i metodi di massima verosimiglianza sono vulnerabili a un gran numero di basi ambigue all’interno dei genomi sequenziati.
Più analisi più estese sono necessarie per valutare l’effetto delle sequenze parziali sulle filogenesi, ma rimuovere le sequenze con > 10% Ns nelle regioni di interesse può essere appropriato in prima istanza.
Le sequenze con sospetti errori di sequenziamento sottostanti (per esempio, indotti da assemblaggi errati) dovrebbero essere studiate e di solito rimosse.
Gli errori di sequenziamento possono manifestarsi come alta divergenza rispetto ad altre sequenze o come un alto numero di sostituzioni in brevi regioni che possono indicare assemblaggi errati locali.
Un alto numero di basi non-ACGTN può essere indicativo di popolazioni virali popolazioni virali miste a seguito di contaminazione.
Sono disponibili diversi strumenti utili per aiutare a rilevare basi ambigue, indel (inserzioni o delezioni di basi) e frame-shift, compresa la funzione Nextclade Quality Control Metric all’interno di Nextstrain (183), CoV-GLUE (188) e Pangolin (189).
Rimozione delle sequenze che formano lunghi rami filogenetici
Le sequenze che formano rami sospettosamente lunghi su un albero filogenetico (che suggeriscono insolitamente alta divergenza evolutiva) dovrebbero essere curate molto attentamente.
Tali rami possono riflettere reali effetti reali, come grandi indel o eventi di ricombinazione, ma nel caso di genomi altamente conservati genomi altamente conservati, tra cui SARS-CoV-2, indicano più comunemente un sostanziale tasso di errore nella sequenza sottostante o un disallineamento (Fig. 4).
Rimuovere le sequenze in cui la divergenza è sostanzialmente maggiore o minore del previsto
Le sequenze sospette possono anche essere identificate utilizzando un albero filogenetico e strumenti come TempEst (190) o TreeTime (191).
In particolare, se una sequenza è sostanzialmente più o meno divergente del previsto dato il tempo in cui è stata campionata, dovrebbe essere attentamente controllata per potenziali errori ed eventualmente rimossa.
Le sequenze che sono più o meno divergenti del previsto possono derivare da problemi bioinformatici (ad esempio, scarsa chiamata delle varianti o taglio inappropriato) o da metadati errata attribuzione, ad esempio una data di campionamento errata.
Esattamente ciò che costituisce “troppo divergente” non è formalmente definito, ma dovrebbero essere indagati i chiari outlier.
L’ispezione manuale per identificare le caratteristiche che possono indicare errori nell’assemblaggio genomico del virus è spesso utile per i set di dati più piccoli.
Tali caratteristiche possono includere inserzioni, sostituzioni o delezioni che portano a codoni di stop all’interno delle sequenze di codifica previste, o brevi stringhe di basi che sono altamente divergenti da tutte le altre sequenze nell’allineamento, in particolare quando i siti vicini hanno chiamate di basi ambigue.
Fig. 4. Ramo lungo spurio mostrato su una filogenesi di massima verosimiglianza non radicata costruita da un allineamento di genomi completi e parziali di SARS-CoV-2, dove un singolo genoma era disallineato rispetto al il resto. Questo è un esempio estremo. Piccoli assemblaggi errati o brevi regioni di disallineamento possono risultare in un ramo terminale che è più lungo della media ma non così estremo.
6.8.4 Rimozione di sequenze ricombinanti
Mentre non ci sono prove, ad oggi, di ricombinazione all’interno della SARS-CoV-2, i coronavirus sono noti per ricombinarsi, e le sequenze dovrebbero essere controllate per forme ricombinanti man mano che la pandemia si espande.
I virus ricombinanti non possono essere adeguatamente collocati all’interno di un albero filogenetico da una singola analisi dell’intero genoma, poiché le sezioni del genoma di ogni virus ancestrale avrebbero storie diverse e sarebbero quindi collocate in posizioni filogenetiche diverse.
L’inclusione di sequenze ricombinanti può portare a stime errate del tasso evolutivo e posizionamento filogenetico.
Se vengono rilevate sequenze ricombinanti, esse possono essere rimosse o diversi alberi filogenetici possono essere stimati dalle sottosezioni dell’allineamento che cadono su entrambi i lati dei punti di rottura della ricombinazione.
Rilevare la ricombinazione è una sfida per molti set di dati sulla SARS-CoV-2 perché gli strumenti esistenti non sono progettati per essere utilizzati su set di dati così grandi, con migliaia di sequenze che hanno anche una diversità genetica relativamente bassa.
Il rilevamento di omoplasie multiple (in cui una sostituzione è sorta indipendentemente in lignaggi filogenetici separati) può indicare la possibilità di una ricombinazione, ma dovrebbe essere attentamente indagato in quanto le omoplasie possono anche essere causate da mutazioni.
Il software RDP4 può essere utilizzato per esaminare fino a 2500 sequenze allineate utilizzando vari test di ricombinazione (192), anche se la sua sensibilità per il rilevamento accurato della ricombinazione all’interno dei lignaggi di SARS-CoV-2 non è ancora stata determinata.
Miglioramenti o strategie di riferimento per il rilevamento della ricombinazione all’interno dei set di dati di SARS-CoV-2 sarebbe vantaggioso.
6.8.5 Strumenti filogenetici
Con un allineamento del genoma di alta qualità, è possibile ricostruire il corrispondente albero filogenetico.
I metodi filogenetici Neighbour-joining sono rapidi e possono essere utili per l’esplorazione iniziale di grandi insiemi di dati genetici.
Tuttavia, essi considerano solo un singolo albero possibile e non dovrebbero essere utilizzati per fare inferenze sulla parentela filogenetica.
Anche FastTree è rapido e produce un stima filogenetica approssimata di massima verosimiglianza, che può essere un’alternativa alternativa ai metodi di neighbour-joining per l’esplorazione dei dati (193).
Molti programmi filogenetici e filodinamici di massima verosimiglianza e bayesiani sono appropriati per l’inferenza filogenetica.
Ognuno richiede la specificazione di un modello di evoluzione del sito.
Questo può essere scelto sulla base delle informazioni contenute nell’allineamento, usando software come ModelTest-NG (195).
Il software comunemente usato per l’inferenza dell’albero di massima verosimiglianza include PhyML (195), RAxML (196) e IQ-TREE (197, 198). RAxML è specificamente progettato per la velocità di esecuzione quando l’allineamento contiene migliaia di sequenze, mentre PhyML e IQ-TREE sono più lenti ma hanno dimostrato costantemente di essere molto accurati.
IQ-TREE ha la funzionalità aggiunta di eseguire prima un test del modello per identificare la scelta più scelta più appropriata del modello di sostituzione dai dati, e fornisce anche un metodo di bootstrapping ultrarapido per stimare il supporto dei rami.
IQ-TREE esegue anche un controllo di complessità sui dati di input e rifiuta le sequenze che contengono troppe ambiguità o altri artefatti che che dovrebbero interferire con la ricostruzione della filogenesi.
Le statistiche di supporto dei rami (ad esempio il supporto da 100 bootstraps, in cui 100 alberi sono rivalutati sulla base di allineamenti fittizi generati da ricampionamento casuale con sostituzione dei siti nei veri siti di allineamento) dovrebbe sempre essere calcolato per valutare la robustezza dei modelli di clustering.
Tali approcci filogenetici sono utili per studiare la parentela evolutiva, ma non possono essere usati per eseguire inferenze filodinamiche inferenze filodinamiche (sezioni 5.4 e 5.5).
Per piccoli insiemi di dati (idealmente non più di 500-1000 per evitare un completamento estremamente lento e problemi di convergenza, anche se il numero esatto dipende dalla disponibilità di calcolo ad alte prestazioni di calcolo ad alte prestazioni e dall’insieme di dati in questione), può essere possibile utilizzare metodi probabilistici come quelli implementati in BEAST (199) o BEAST2 (200).
Questi metodi possono essere utilizzati per stimare il tempo di comparsa di particolari cladi di interesse (ad esempio, focolai locali), la diffusione geografica di un focolaio, e i parametri demografici, compresa la dimensione della popolazione del virus nel tempo (sezioni 5.4 e 5.5).
Per le analisi incentrate esclusivamente sulla stima del tempo trascorso dalla divergenza per un gruppo di genomi virali, specialmente quando questi set di dati sono grandi, può essere sufficiente e più computazionalmente trattabile utilizzare metodi meno complessi che combinano le date di campionamento con alberi di massima verosimiglianza precompilati, come i programmi software least-squares dating (LSD) (201) o TreeTime (191).
Tutti questi metodi richiedono un “segnale temporale” sufficiente all’interno del set di dati, in modo che si possa vedere che i lignaggi del virus si evolvono in modo simile a un orologio con sostituzioni che avvengono a un ritmo relativamente prevedibile.
Esattamente come tracciare la linea di demarcazione tra un segnale temporale insufficiente e sufficiente rispetto alla SARS-CoV-2 è stato l’obiettivo di gran parte del lavoro filodinamico iniziale (45).
Ora ci sono stati molteplici esempi di analisi filogenetiche e filodinamiche a scala temporale della SARS-CoV-2 (59, 85).
Mentre la soglia filodinamica (il punto nel tempo in cui si è accumulato un cambiamento evolutivo molecolare sufficiente accumulati nei campioni di genoma disponibili per ottenere stime filodinamiche robuste) è stata raggiunto per alcune analisi, sottoinsiemi dei dati di sequenza disponibili corrispondenti a cluster locali all’interno di specifiche aree geografiche dovrebbero essere trattati con cura e rivalutati prima dell’uso, al fine di al fine di determinare l’applicabilità dei metodi filodinamici.
Mentre i metodi basati sulle reti (ad esempio i metodi di unione degli aplotipi, le reti di giunzione mediana) sono sono rapidi e semplici da eseguire e sono presenti nella letteratura pubblicata sulla SARS-CoV-2, le reti mancano di un adeguato radicamento filogenetico che è importante per la comprensione delle storie storie evolutive.
Mancano anche di un modello appropriato di evoluzione del sito, essendo basati invece sulla somiglianza delle sole sequenze del genoma, e non valutano o catturano la robustezza dei modelli di connettività visualizzati modelli.
La costruzione di un albero filogenetico sarà quindi solitamente altrettanto appropriata, o più appropriata, rispetto alla costruzione di una rete per analizzare le sequenze del genoma virale della SARS-CoV-2 (202).
6.8.6 Visualizzazione
Gli alberi filogenetici possono essere visualizzati localmente usando un’ampia varietà di software disponibili gratuitamente (per esempio FigTree e MEGA (203)) e commerciali.
L’applicazione web Microreact fornisce una visualizzazione interattiva di un albero filogenetico inserito dall’utente, permettendo la strutturazione filogenetica per località (longitudine e latitudine), categoria (ad esempio paese) e tempo da visualizzare (204).
La mappatura delle posizioni delle punte filogenetiche relative all’albero posizione dell’albero può essere utile per esplorare la strutturazione geografica della diversità della SARS-CoV-2, e per rapida conferma, se del caso, che i dati siano stati geocodificati correttamente.
Microreact richiede un file di input contenente metadati, come la data e la posizione del campionamento, e un albero filogenetico.
I progetti caricati possono essere condivisi pubblicamente o mantenuti privati, e aggiornati dall’utente come richiesto. utente come richiesto.
I progetti attualmente disponibili e accessibili pubblicamente includono una distribuzione globale di SARS-CoV-2 che viene aggiornata dal COVID-19 Genomics UK Consortium.
I file degli alberi filogenetici non sono mostrati con le statistiche di supporto dei rami, e quindi gli alberi da quindi gli alberi dei set di dati pubblicamente disponibili devono essere scaricati per l’ispezione locale, se necessario.
Ulteriori informazioni aggiuntive sui metodi usati per costruire le filogenesi sono fornite a discrezione dell’autore del progetto. a discrezione dell’autore del progetto; ciò è utile per consentire un’adeguata considerazione di queste filogenesi.
Nextstrain (183) fornisce una visualizzazione interattiva dell’evoluzione e della diversità geografica di SARS-CoV-2 e di altri patogeni.
I collaboratori e gli sviluppatori curano visualizzazioni filogenetiche globali e regionali visualizzazioni filogenetiche online che sono state frequentemente consultate durante la pandemia COVID-19.
Gli utenti possono impostare la propria filogenesi Augur locale e le visualizzazioni delle mappe per analizzare dati basati su file di input di sequenze, filogenesi e metadati.
Nextstrain è uno strumento potente e uno strumento potente e rapido per esplorare modelli su larga scala di strutturazione geografica.
Tuttavia, qualsiasi filogenesi dovrebbe sempre essere interpretata con cautela, prendendo in considerazione gli intervalli di confidenza intervalli di fiducia nelle date di divergenza fornite e l’incertezza nella posizione geografica visualizzata, e nel contesto delle “Nextstrain narratives” esplicative, se disponibili.
Gli alberi filogenetici non sono visualizzati con le statistiche di supporto dei rami, quindi l’ordine di ramificazione mostrato non dovrebbe essere assunto per essere esatto o utilizzato per informare le decisioni politiche senza ulteriori indagini per confermare i risultati.
Le posizioni geografiche e i tempi di divergenza dei rami filogenetici sono dedotti utilizzando metodi meno complessi ma più rapidi di quelli comunemente impiegati in BEAST o BEAST2.
Le analisi che confrontano il grado di accordo tra i diversi metodi per SARS-CoV-2 sarebbero preziose: le disparità tra i diversi metodi non sono rare (205).
Come descritto nella sezione 5, il campionamento non casuale delle sequenze può falsare le interpretazioni filogenetiche e interpretazioni e conclusioni filogenetiche e filogeografiche. È importante essere consapevoli di questi possibili distorsioni quando si interpretano le visualizzazioni filogenetiche.
6.8.7 Classificazione del lignaggio
Attualmente non esiste un sistema di denominazione formale universalmente accettato per i lignaggi evolutivi della SARS-CoV-2.
Diverse nomenclature proposte usano gli stessi nomi (ad esempio “A1”) per riferirsi a diverse e quindi è importante dichiarare quale nomenclatura viene utilizzata in ogni descrizione.
L’adozione globale di un unico sistema di nomenclatura faciliterebbe la comunicazione scientifica comunicazione scientifica su lignaggi specifici ed eviterebbe la confusione generata dall’uso di più sistemi.
Attualmente ci sono tre sistemi di nomenclatura comunemente usati per la SARS-CoV-2 cladi/linee.
Sia GISAID EpiCoV™ che Nextstrain mirano a fornire un’ampia categorizzazione della diversità circolante a livello globale attraverso la denominazione di diversi cladi filogenetici.
Rambaut et al. (189) hanno proposto una nomenclatura dinamica per i lignaggi della SARS-CoV-2 che si concentra sui lignaggi del virus che circolano attivamente e su quelli che si diffondono in nuove località.
È disponibile un software che permette agli utenti di assegnare le proprie sequenze a questi lignaggi, anche tramite Pangolin, Nextstrain e CoV-Glue (183, 188, 189).
Dato che attualmente non esiste una nomenclatura universalmente accettata, l’approccio migliore quando si riportano i lignaggi è quello di dichiarare la nomenclatura di particolari cladi in tutti e tre i sistemi comunemente usati, o almeno di indicare esplicitamente quale nomenclatura viene utilizzata.
6.8.8 Radicamento filogenetico
Indipendentemente dal software filogenetico e dal metodo usato, la scelta di uno o più outgroups è importante e avrà un effetto sulla determinazione della radice dell’albero. importante e avrà un effetto su come viene determinata la radice dell’albero.
Questo a sua volta influenzare le stime del tempo dalla divergenza.
Un outgroup è una sequenza selezionata per essere il più correlata il più possibile alle sequenze di interesse, ma nota per non essere parte dello stesso clade.
In pratica, la prima sequenza di riferimento disponibile di SARS-CoV-2 è spesso usata come outgroup quando si costruisce una filogenesi di genomi provenienti da una varietà di fonti geografiche.
Per l’indagine dei cluster locali, può essere opportuno scegliere un genoma più strettamente correlato dall’esterno dell’insieme di dati da analizzare.
7. Conclusioni e necessità future
Il sequenziamento rapido dei genomi dei virus è ora possibile in vari contesti, e le analisi delle sequenze genomiche di SARS-CoV-2 hanno un enorme potenziale per informare gli sforzi di salute pubblica che circondano COVID-19.
La rapida generazione e la condivisione globale delle sequenze genomiche dei virus fornisce informazioni che contribuiranno alla comprensione della trasmissione e alla progettazione di strategie cliniche e strategie di mitigazione clinica ed epidemiologica.
Il dialogo tra enti di salute pubblica, generatori di dati e analisti è fondamentale per garantire che i dati siano generati e usati in modo appropriato per il massimo beneficio per la salute pubblica.
Un’attenta considerazione preventiva considerazione del motivo per cui il sequenziamento viene condotto è necessario, in quanto ciò influenzerà la scelta dei campioni, la raccolta di metadati e le successive analisi.
Il sequenziamento dovrebbe essere condotto con la dovuta considerazione delle risorse e delle capacità disponibili, e non dovrebbe sottrarre capacità da altre aree ugualmente vitali.
Dovrebbero essere stabiliti chiari canali di comunicazione per condividere risultati, campioni e dati con le parti interessate, in modo che le informazioni possano essere utilizzate per migliorare la salute pubblica il più rapidamente possibile.
Tradurre le sequenze del genoma della SARS-CoV-2 in risultati informativi è complesso e spesso richiede una formazione specialistica sostanziale per assicurare che le violazioni dei presupposti del modello non portino una comprensione errata dell’epidemiologia del virus.
Una chiara comprensione dei benefici e dei limiti delle analisi genomiche permetterà una valutazione sicura di dove gli strumenti genomici possono aumentare o sostenere gli approcci esistenti e dove, al contrario, la modellazione epidemiologica o sperimentazione di laboratorio può essere più solida.
La collaborazione tra esperti con diverse abilità è preziosa, poiché non tutti i laboratori hanno competenze locali esistenti in tutti i settori.
Nonostante i recenti progressi nella facilità con cui si possono generare sequenze di virus, le sfide rimangono.
In molti contesti, la necessità di una rapida importazione di reagenti sensibili alla temperatura è stata una ostacolo significativo all’adozione di approcci di sequenziamento portatili all’interno del paese all’inizio durante durante il COVID-19.
Le soluzioni devono essere trovate se i paesi devono sviluppare la loro capacità di condurre attività di sequenziamento in future emergenze di salute pubblica così come durante l’attuale pandemia.
I finanziamenti che sostengono le attività per convalidare e confrontare le diverse strategie di strategie di sequenziamento e analisi sarebbero anche molto utili per assicurare un’appropriata selezione.
L’analisi e l’interpretazione dei dati di sequenza genomica dei virus non sono semplici.
I laboratori che pianificano di adottare il sequenziamento per la prima volta potrebbero beneficiare di programmi che forniscono supporto per la convalida formale delle loro pipeline di sequenziamento.
I dati genomici globali generati per la SARS-CoV-2 sono troppo grandi per molti strumenti attuali ; sono necessari miglioramenti per permettere di analizzare rapidamente serie di dati sempre più grandi durante le emergenze di salute pubblica e, dove possibile, per aumentare il livello di automazione.
Una migliore comprensione accademica di ciò che agenzie di salute pubblica e di come i risultati possono essere presentati al meglio per enfatizzare le implicazioni pratiche, pur tenendo conto dell’incertezza analitica, sarebbe anche utile.
I laboratori di salute pubblica hanno generalmente più esperienza nella genetica molecolare che nella filogenetica computazionale e bioinformatica. Un investimento rafforzato e a lungo termine in formazione in filogenetica e bioinformatica è necessario per ottenere il massimo beneficio dalla crescita delle possibilità di sequenziamento in laboratorio in questa e nelle successive epidemie.
Repository come GISAID hanno incoraggiato e facilitato la condivisione dei dati su COVID-19.
Tuttavia, sono ancora necessarie discussioni più ampie per assicurare continui miglioramenti nella condivisione dei dati durante le emergenze di salute pubblica.
Attualmente, molti ricercatori rimangono riluttanti a condividere i dati di sequenza genomica genomica fino a quando non è stata preparata una pubblicazione pre-print.
Le ragioni di questo dovrebbero essere cercare e proporre soluzioni.
Una discussione più ampia e un accordo sull’appropriato accreditamento per i produttori di dati in diverse circostanze è anche necessario per incoraggiare la la condivisione dei dati.
C’è bisogno di sviluppare nuovi standard o metriche di accreditamento dei dati e che le riviste si impegnino a sostenere pratiche eque di utilizzo dei dati.
Un più ampio impegno pubblico da parte degli scienziati è importante per ridurre la diffusione di false informazioni false durante le emergenze sanitarie attuali e future.
Maggiore sostegno e formazione per gli scienziati su come i messaggi scientifici possono essere efficacemente condivisi con il pubblico sarebbe utile. Assicurarsi che i pazienti e il pubblico comprendano il valore e i limiti dei dati sulle sequenze genomiche dei virus è essenziale per sostenere le consultazioni pubbliche sull uso appropriato dei metadati dei pazienti durante le emergenze di salute pubblica.
Bibliografia
1. Roy S, LaFramboise WA, Nikiforov YE, Nikiforova MN, Routbort MJ, Pfeifer J et al. Next-generation sequencing informatics: challenges and strategies for implementation in a clinical environment. Arch Pathol Lab Med. 2016;140:958-75. doi: 10.5858/arpa.2015-0507-RA.
2. Gu W, Miller S, Chiu CY. Clinical metagenomic next-generation sequencing for pathogen detection. Annu Rev Pathol. 2019;14:319-38. doi: 10.1146/annurev-pathmechdis-012418-012751.
3. Houldcroft CJ, Beale MA, Breuer J. Clinical and biological insights from viral genome sequencing. Nat Rev Microbiol. 2017;15:183-92. doi: 10.1038/nrmicro.2016.182.
4. Quick J, Loman NJ, Duraffour S, Simpson JT, Severi E, Cowley L et al. Real-time, portable genome sequencing for Ebola surveillance. Nature. 2016;530:228-32. doi: 10.1038/nature16996.
5. Peiris JSM, Lai ST, Poon LLM, Guan Y, Yam LYC, Lim W et al. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet. 2003;361:1319-25. doi: 10.1016/s0140-6736(03)13077-2.
6. Drosten C, Günther S, Preiser W, van der Werf S, Brodt H-R, Becker S et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Eng J Med. 2003;348:1967-76. doi: 10.1056/NEJMoa030747.
7. Ksiazek TG, Erdman D, Goldsmith CS, Zaki SR, Peret T, Emery S et al. A novel coronavirus associated with severe acute respiratory syndrome. N Eng J Med. 2003;348:1953-66. doi: 10.1056/NEJMoa030781.
8. Zhu N, Zhang D, Wang W, Li X, Yang B, Song J et al. A novel coronavirus from patients with pneumonia in China, 2019. N Eng J Med. 2020;382:727-33. doi: 10.1056/NEJMoa2001017.
9. World Health Organization. Novel coronavirus (2019-nCoV): Situation report 1. Geneva ; 2020 (sitrep-1-2019-ncov.pdf?sfvrsn=20a99c10_4, accessed 2 November 2020).
10. Fraser C, Donnelly CA, Cauchemez S, Hanage WP, Van Kerkhove MD, Hollingsworth TD et al. Pandemic potential of a strain of influenza A(H1N1): early findings. Science. 2009;324:1557-61. doi: 10.1126/science.1176062.
11. Rambaut A, Holmes E. The early molecular epidemiology of the swine-origin A/H1N1 human influenza pandemic. PLoS Curr. 2009;1:RRN1003. doi: 10.1371/currents.rrn1003.
12. Smith GJD, Vijaykrishna D, Bahl J, Lycett SJ, Worobey M, Pybus OG et al. Origins and evolutionary genomics of the 2009 swine-origin H1N1 influenza A epidemic. Nature. 2009;459:1122-5. doi: 10.1038/nature08182.
13. Mena I, Nelson MI, Quezada-Monroy F, Dutta J, Cortes-Fernández R, Lara-Puente JH et al. Origins of the 2009 H1N1 influenza pandemic in swine in Mexico. eLife. 2016;5:e16777. doi: 10.7554/eLife.16777.
14. WHO MERS-CoV Research Group. State of knowledge and data gaps of Middle East respiratory syndrome coronavirus (MERS-CoV) in humans. PLoS Curr. 2013;5. doi: 10.1371/currents.outbreaks.0bf719e352e7478f8ad85fa30127ddb8.
15. Haagmans BL, Al Dhahiry SHS, Reusken CBEM, Raj VS, Galiano M, Myers R et al. Middle East respiratory syndrome coronavirus in dromedary camels: an outbreak investigation. Lancet Infect Dis. 2014;14:140-5. doi: 10.1016/S1473-3099(13)70690-
16. Sabir JSM, Lam TTY, Ahmed MMM, Li L, Shen Y, Abo-Aba SEM et al. Co-circulation of three camel coronavirus species and recombination of MERS-CoVs in Saudi Arabia. Science. 2016;351:81-4. doi: 10.1126/science.aac8608.
17. Azhar EI, El-Kafrawy SA, Farraj SA, Hassan AM, Al-Saeed MS, Hashem AM et al. Evidence for camel-to-human transmission of MERS coronavirus. N Eng J Med. 2014;370:2499-505. doi: 10.1056/NEJMoa1401505.
18. Memish ZA, Cotten M, Meyer B, Watson SJ, Alsahafi AJ, Al Rabeeah AA et al. Human infection with MERS coronavirus after exposure to infected camels, Saudi Arabia, 2013. Emerg Infect Dis. 2014;20:1012-5. doi: 10.3201/eid2006.140402.
19. Chu DKW, Hui KPY, Perera RAPM, Miguel E, Niemeyer D, Zhao J et al. MERS coronaviruses from camels in Africa exhibit region-dependent genetic diversity. Proc Natl Acad Sci USA. 2018;115:3144-9. doi: 10.1073/pnas.1718769115.
20. Dudas G, Carvalho LM, Rambaut A, Bedford T. MERS-CoV spillover at the camel-human interface. eLife. 2018;7:e31257. doi: 10.7554/eLife.31257. 21. Baize S, Pannetier D, Oestereich L, Rieger T, Koivogui L, Magassouba NF et al. Emergence of Zaire Ebola virus disease in Guinea. N Eng J Med. 2014;371:1418-25. doi: 10.1056/NEJMoa1404505.
22. Gire SK, Goba A, Andersen KG, Sealfon RSG, Park DJ, Kanneh L et al. Genomic surveillance elucidates Ebola virus origin and transmission during the 2014 outbreak. Science. 2014;345:1369-72. doi: 10.1126/science.1259657.
23. Carroll MW, Matthews DA, Hiscox JA, Elmore MJ, Pollakis G, Rambaut A et al. Temporal and spatial analysis of the 2014–2015 Ebola virus outbreak in West Africa. Nature. 2015;524:97-101. doi: 10.1038/nature14594.
24. Dudas G, Rambaut A. Phylogenetic analysis of Guinea 2014 ebov ebolavirus outbreak. PLoS Curr. 2014;6. doi:10.1371/currents.outbreaks.84eefe5ce43ec9dc0bf0670f7b8b417d.
25. Park DJ, Dudas G, Wohl S, Goba A, Whitmer SLM, Andersen KG et al. Ebola virus epidemiology, transmission, and evolution during seven months in Sierra Leone. Cell. 2015;161:1516-26. doi: 10.1016/j.cell.2015.06.007.
26. Simon-Loriere E, Faye O, Faye O, Koivogui L, Magassouba N, Keita S et al. Distinct lineages of Ebola virus in Guinea during the 2014 West African epidemic. Nature. 2015;524:102-4. doi: 10.1038/nature14612.
27. Tong Y-G, Shi W-F, Liu D, Qian J, Liang L, Bo X-C et al. Genetic diversity and evolutionary dynamics of Ebola virus in Sierra Leone. Nature. 2015;524:93-6. doi: 10.1038/nature14490.
28. Ladner JT, Wiley MR, Mate S, Dudas G, Prieto K, Lovett S et al. Evolution and spread of Ebola virus in Liberia, 2014-2015. Cell Host Microbe. 2015;18:659-69. doi: 10.1016/j.chom.2015.11.008.
29. Volz E, Pond S. Phylodynamic analysis of Ebola virus in the 2014 Sierra Leone epidemic. PLOS Curr. 2014;24 doi:10.1371/currents.outbreaks.6f7025f1271821d4c815385b08f5f80e.
30. Stadler T, Kühnert D, Rasmussen DA, Plessis DL. Insights into the early epidemic spread of Ebola in Sierra Leone provided by viral sequence data. PLOS Curr. 2014. doi: 10.1371/currents.outbreaks.02bc6d927ecee7bbd33532ec8ba6a25f.
31. Mate SE, Kugelman JR, Nyenswah TG, Ladner JT, Wiley MR, Cordier-Lassalle T et al. Molecular evidence of sexual transmission of Ebola virus. N Eng J Med. 2015;373:2448-54. doi: 10.1056/NEJMoa1509773.
32. Felsenstein J. Cases in which parsimony or compatibility methods will be positively misleading. Syst Zoology. 1978;27:401-10. doi: 10.2307/2412923.
33. Holmes EC, Dudas G, Rambaut A, Andersen KG. The evolution of Ebola virus: Insights from the 2013–2016 epidemic. Nature. 2016;538:193-200. doi: 10.1038/nature19790.
34. Arias A, Watson SJ, Asogun D, Tobin EA, Lu J, Phan MVT et al. Rapid outbreak sequencing of Ebola virus in Sierra Leone identifies transmission chains linked to sporadic cases. Vir Evol. 2016;2:vew016. doi: 10.1093/ve/vew016.
35. Hoenen T, Groseth A, Rosenke K, Fischer RJ, Hoenen A, Judson SD et al. Nanopore sequencing as a rapidly deployable Ebola outbreak tool. Emerg Infect Dis. 2016;22:331-4. doi: 10.3201/eid2202.151796.
36. Smits SL, Pas SD, Reusken CB, Haagmans BL, Pertile P, Cancedda C et al. Genotypic anomaly in Ebola virus strains circulating in Magazine Wharf area, Freetown, Sierra Leone, 2015. Euro Surveill. 2015;20. doi: 10.2807/1560-7917.ES.2015.20.40.30035.
37. Faria NR, Quick J, Claro IM, Thézé J, de Jesus JG, Giovanetti M et al. Establishment and cryptic transmission of Zika virus in Brazil and the Americas. Nature. 2017;546:406-10. doi: 10.1038/nature22401.
38. Faria NR, Azevedo RdSdS, Kraemer MUG, Souza R, Cunha MS, Hill SC et al. Zika virus in the Americas: early epidemiological and genetic findings. Science. 2016;352:345-9. doi: 10.1126/science.aaf5036.
39. Metsky HC, Matranga CB, Wohl S, Schaffner SF, Freije CA, Winnicki SM et al. Zika virus evolution and spread in the Americas. Nature. 2017;546:411-5. doi: 10.1038/nature22402.
40. Grubaugh ND, Ladner JT, Kraemer MUG, Dudas G, Tan AL, Gangavarapu K et al. Genomic epidemiology reveals multiple introductions of zika virus into the United States. Nature. 2017;546:401-5. doi: 10.1038/nature22400.
41. Grubaugh ND, Ladner JT, Lemey P, Pybus OG, Rambaut A, Holmes EC et al. Tracking virus outbreaks in the twenty-first century. Nat Microbiol. 2019;4:10. doi: 10.1038/s41564-018-0296-2.
42. Gardy JL, Loman NJ. Towards a genomics-informed, real-time, global pathogen surveillance system. Nat Rev Genet. 2018;19:9-20. doi: 10.1038/nrg.2017.88.
43. Rasmussen AL, Katze MG. Genomic signatures of emerging viruses: a new era of systems epidemiology. Cell Host Microbe. 2016;19:611-8. doi: 10.1016/j.chom.2016.04.016.
44. Loewe L, Hill WG. The population genetics of mutations: Good, bad and indifferent. Philos Trans R Soc Lond B Biol Sci. 2010;365:1153-67. doi: 10.1098/rstb.2009.0317.
45. Duchene S, Featherstone L, Haritopoulou-Sinanidou M, Rambaut A, Lemey P, Baele G. Temporal signal and the phylodynamic threshold of SARS-CoV-2. Virus Evol. 2020: 19;6(2). doi:10.1093/ve/veaa061.
46. Duffy S, Shackelton LA, Holmes EC. Rates of evolutionary change in viruses: patterns and determinants. Nat Rev Genet. 2008;9:267-76. doi: 10.1038/nrg2323.
47. Grenfell BT, Pybus OG, Gog JR, Wood JLN, Daly JM, Mumford JA et al. Unifying the epidemiological and evolutionary dynamics of pathogens. Science. 2004;303:327-32. doi: 10.1126/science.1090727.
49. Pybus OG, Rambaut A. Evolutionary analysis of the dynamics of viral infectious disease. Nat Rev Genet. 2009;10:540-50. doi: 10.1038/nrg2583.
50. Sanjuán R, Domingo-Calap P. Mechanisms of viral mutation. Cell Mol Life Sci.2016;73:4433-48. doi: 10.1007/s00018-016-2299-6.
51. Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. The species severe acute respiratory syndrome-related coronavirus : classifying 2019-nCov and naming it SARS-CoV-2. Nat Microbiol. 2020;5:536-44. doi: 10.1038/s41564-020-0695-z.
52. Wu F, Zhao S, Yu B, Chen Y-M, Wang W, Song Z-G et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265-9. doi: 10.1038/s41586-020-2008-3.
53. Candido DDS, Claro IM, Jesus DJG, Souza DWM, Moreira FRR, Dellicour S et al. Evolution and epidemic spread of SARS-CoV-2 in Brazil. Science (New York, NY). 2020;369:1255-60. doi: 10.1101/2020.06.11.20128249.
54. Emanuel EJ, Wendler D, Grady C. What makes clinical research ethical? JAMA. 2000; 283(20): 2701-11.
55. WHO guidelines on ethical issues in public health surveillance. Geneva: World Health Organization; 2017 (surveillance/en/, accessed 15 November 2020).
56. World Health Organization. Policy statement on data sharing by the World Health Organization in the context of public health emergencies. Geneva; 2016.
57. Thézé J, Li T, Plessis dL, Bouquet J, Kraemer MUG, Somasekar S et al. Genomic epidemiology reconstructs the introduction and spread of Zika virus in Central America and Mexico. Cell Host Microbe. 2018;23:855-64.e7. doi: 10.1016/j.chom.2018.04.017.
58. COVID-19 data portal. 2020 (https://www.covid19dataportal.org/sequences, accessed 1 November 2020).
59. Lu J, du Plessis L, Liu Z, Hill V, Kang M, Lin H et al. Genomic epidemiology of SARS-CoV-2 in Guangdong Province, China. Cell. 2020;181:997-1003.e9. doi: 10.1016/j.cell.2020.04.023.
60. Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270-3. doi: 10.1038/s41586-020-2012-7.
61. Li Q, Guan X, Wu P, Wang X, Zhou L, Tong Y et al. Early transmission dynamics in Wuhan, China, of novel coronavirus-infected pneumonia. N Engl J Med. 2020;382:1199-207. doi: 10.1056/NEJMoa2001316.
62. Andersen KG, Rambaut A, Lipkin WI, Holmes EC, Garry RF. The proximal origin of SARS-CovV2. Nature Medicine. 2020;26:450-2. doi: 10.1038/s41591-020-0820-9.
63. Report of the WHO-China Joint Mission on Coronavirus Disease 2019 (COVID-19). Geneva: World Health Organization; 2020.(https://www.who.int/publications/i/item/report-of-the-who-china-joint-mission-on-coronavirus-disease-2019-(covid-19), accessed 28 December 2020)
64. Cui J, Li F, Shi Z-L. Origin and evolution of pathogenic coronaviruses. Nat Rev Microbiol. 2019;17:181-92. doi: 10.1038/s41579-018-0118-9.
65. Hu B, Zeng L-P, Yang X-L, Ge X-Y, Zhang W, Li B et al. Discovery of a rich gene pool of bat SARS-related coronaviruses provides new insights into the origin of SARS coronavirus. PLoS Pathog. 2017;13:e1006698. doi: 10.1371/journal.ppat.1006698.
66. Lin X-D, Wang W, Hao Z-Y, Wang Z-X, Guo W-P, Guan X-Q et al. Extensive diversity of coronaviruses in bats from China. Virology. 2017;507:1-10. doi: 10.1016/j.virol.2017.03.019.
67. Boni MF, Lemey P, Jiang X, Lam TT-Y, Perry B, Castoe T et al. Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic. Nature Microbiology. 2020;5:1408-17. doi: 10.1101/2020.03.30.015008.
68. Zhou H, Chen X, Hu T, Li J, Song H, Liu Y et al. A novel bat coronavirus closely related to SARS-CoV-2 contains natural insertions at the S1/S2 cleavage site of the spike protein. Curr Biol. 2020;30:2196-203.e3. doi: 10.1016/j.cub.2020.05.023.
69. Lam TT-Y, Jia N, Zhang Y-W, Shum MH-H, Jiang J-F, Zhu H-C et al. Identifying SARS-CoV-2-related coronaviruses in Malayan pangolins. Nature. 2020:1-4. doi: 10.1038/s41586-020-2169-0.
70. Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271-80.e8. doi: 10.1016/j.cell.2020.02.052.
71. Letko M, Marzi A, Munster V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat Microbiol. 2020;5:562-9. doi: 10.1038/s41564-020-0688-y.
72. Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh C-L, Abiona O et al. Cryo-em structure of the 2019-nCoV spike in the prefusion conformation. Science (New York, Ny). 2020;367:1260-3. doi: 10.1126/science.abb2507.
73. Colson P, Scola BL, Esteves-Vieira V, Ninove L, Zandotti C, Jimeno M-T et al. Plenty of coronaviruses but no SARS-CoV-2. (Letter to the editor). Euro Surveill. 2020;25:2000171. doi: 10.2807/1560-7917.ES.2020.25.8.2000171.
74. Corman VM, Landt O, Kaiser M, Molenkamp R, Meijer A, Chu DK et al. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Euro Surveill. 2020;25. doi: 10.2807/1560-7917.ES.2020.25.3.2000045.
75. Information for laboratories about coronavirus (COVID-19). Atlanta: Centers for Disease Control and Prevention; 2020. (panel-primer-probes.html, accessed 26 June 2020).
76. University of Hong Kong, School of Public Health. Detection of 2019 novel coronavirus (2019-nCoV) in suspected human cases by RT-PCR. 2020 (20.pdf?sfvrsn=af1aac73_4.
77. Diagnostic testing for SARS-CoV-2. Interim guidance. Geneva: World Health Organization; 2020 (cov-2, accessed 19 November 2020).
78. Ren Y, Zhou Z, Liu J, Lin L, Li S, Wang H et al. A strategy for searching antigenic regions in the SARS-CoV spike protein. Genomics Proteomics Bioinformatics. 2003;1:207-15. doi: 10.1016/s1672-0229(03)01026-x.
79. Kumar S, Maurya VK, Prasad AK, Bhatt MLB, Saxena SK. Structural, glycosylation and antigenic variation between 2019 novel coronavirus (2019-nCoV) and SARS coronavirus (SARS-CoV). Virusdisease. 2020:1-9. doi: 10.1007/s13337-020-00571-5.
80. Melén K, Kakkola L, He F, Airenne K, Vapalahti O, Karlberg H et al. Production, purification and immunogenicity of recombinant Ebola virus proteins – a comparison of Freund’s adjuvant and adjuvant system 03. J Virol Methods. 2017;242:35-45. doi:10.1016/j.jviromet.2016.12.014.
81. Ziegler T, Matikainen S, Rönkkö E, Österlund P, Sillanpää M, Sirén J et al. Severe acute respiratory syndrome coronavirus fails to activate cytokine-mediated innate immune responses in cultured human monocyte-derived dendritic cells. J Virol. 2005;79:13800-5. doi: 10.1128/JVI.79.21.13800-13805.2005.
82. Draft landscape of COVID-19 candidate vaccines. Geneva: World Health Organization; 2020 (vaccines, accessed 26 June 2020).
83. Li G, Clercq ED. Therapeutic options for the 2019 novel coronavirus (2019-nCoV). Nat Rev Drug Discov. 2020;19:149-50. doi: 10.1038/d41573-020-00016-0.
84. Bedford T, Greninger AL, Roychoudhury P, Starita LM, Famulare M, Huang M-L et al. Cryptic transmission of SARS-CoV-2 in Washington State. Science. 2020;370:571-5. doi: 10.1101/2020.04.02.20051417.
85. Volz E, Fu H, Wang H, Xi X, Chen W, Liu D et al. Genomic epidemiology of a densely sampled COVID19 outbreak in China. medRxiv. 2020:2020.03.09.20033365. doi: 10.1101/2020.03.09.20033365.
86. Zehender G, Lai A, Bergna A, Meroni L, Riva A, Balotta C et al. Genomic characterization and phylogenetic analysis of SARS-CoV-2 in Italy. J Med Virol. 2020;92(9):1637-1640. doi: 10.1002/jmv.25794.
87. Worobey M, Pekar J, Larsen BB, Nelson MI, Hill V, Joy JB et al. The emergence of SARS-CoV-2 in Europe and North America. Science. 2020;370:564-70.
88. Lemey P, Rambaut A, Drummond AJ, Suchard MA. Bayesian phylogeography finds its roots. PLoS Comput Biol. 2009;5:e1000520. doi: 10.1371/journal.pcbi.1000520.
89. Lemey P, Rambaut A, Welch JJ, Suchard MA. Phylogeography takes a relaxed random walk in continuous space and time. Mol Biol Evol. 2010;27:1877-85. doi: 10.1093/molbev/msq067.
90. Bloomquist EW, Lemey P, Suchard MA. Three roads diverged? Routes to phylogeographic inference. Trends Ecol Evol. 2010;25:626-32. doi: 10.1016/j.tree.2010.08.010.
91. Faria NR, Suchard MA, Rambaut A, Lemey P. Towards a quantitative understanding of viral phylogeography. Curr OpinVirol. 2011;1:423-9. doi: 10.1016/j.coviro.2011.10.003.
92. Fauver JR, Petrone ME, Hodcroft EB, Shioda K, Ehrlich HY, Watts AG et al. Coast-to-coast spread of SARS-CoV-2 during the early epidemic in the United States. Cell. 2020;181:990-6.e5. doi: 10.1016/j.cell.2020.04.021.
93. Eden J-S, Rockett R, Carter I, Rahman H, de Ligt J, Hadfield J et al. An emergent clade of SARS-CoV-2 linked to returned travellers from Iran. Virus Evol. 2020;6. doi: 10.1093/ve/veaa027.
94. Lemey P, Hong S, Hill V, Baele G, Poletto C, Colizza V et al. Accommodating individual travel history, global mobility, and unsampled diversity in phylogeography: a SARS-CoV-2 case study. bioRxiv. 2020:2020.06.22.165464. doi: 10.1101/2020.06.22.165464.
95. Ewing G, Rodrigo A. Estimating population parameters using the structured serial coalescent with Bayesian mcmc inference when some demes are hidden. Evol Bioinform Online. 2006;2:117693430600200026. doi: 10.1177/117693430600200026.
96. Maio ND, Wu C-H, O’Reilly KM, Wilson D. New routes to phylogeography: A Bayesian structured coalescent approximation. PLoS Genet. 2015;11:e1005421. doi: 10.1371/journal.pgen.1005421.
97. Lemey P, Rambaut A, Bedford T, Faria N, Bielejec F, Baele G et al. Unifying viral genetics and human transportation data to predict the global transmission dynamics of human influenza H3N2. PLoS Pathog. 2014;10:e1003932. doi: 10.1371/journal.ppat.1003932. ù
98. Chaillon A, Gianella S, Dellicour S, Rawlings SA, Schlub TE, Oliveira MFD et al. HIV persists throughout deep tissues with repopulation from multiple anatomical sources. The J Clin Invest. 2020;130:1699-712. doi: 10.1172/JCI134815.
99. Kalkauskas A, Perron U, Sun Y, Goldman N, Baele G, Guindon S et al. Sampling bias and model choice in continuous phylogeography: getting lost on a random walk. bioRxiv. 2020:2020.02.18.954057. doi: 10.1101/2020.02.18.954057.
100. Nylinder S, Lemey P, De Bruyn M, Suchard MA, Pfeil BE, Walsh N et al. On the biogeography of centipeda: a species-tree diffusion approach. Syst Biol. 2014;63:178-91. doi: 10.1093/sysbio/syt102.
101. Dellicour S, Lemey P, Artois J, Lam TT, Fusaro A, Monne I et al. Incorporating heterogeneous sampling probabilities in continuous phylogeographic inference — application to H5N1 spread in the Mekong region. Bioinformatics. 2020;36:2098-104. doi: 10.1093/bioinformatics/btz882.
102. Dudas G, Carvalho LM, Bedford T, Tatem AJ, Baele G, Faria NR et al. Virus genomes reveal factors that spread and sustained the Ebola epidemic. Nature. 2017;544:309-15. doi: 10.1038/nature22040.
103. Dellicour S, Baele G, Dudas G, Faria NR, Pybus OG, Suchard MA et al. Phylodynamic assessment of intervention strategies for the West African Ebola virus outbreak. Nature Communications. 2018;9:1-9. doi: 10.1038/s41467-018-03763-2.
104. Bielejec F, Lemey P, Baele G, Rambaut A, Suchard MA. Inferring heterogeneous evolutionary processes through time: from sequence substitution to phylogeography. Syst Biol. 2014;63:493-504. doi: 10.1093/sysbio/syu015.
105. Sit THC, Brackman CJ, Ip SM, Tam KWS, Law PYT, To EMW et al. Infection of dogs with SARS-CoV-2. Nature. 2020. doi: 10.1038/s41586-020-2334-5.
106. Oreshkova N, Molenaar RJ, Vreman S, Harders F, Munnink BBO, Honing RWH et al. SARS-CoV-2 infection in farmed minks, the Netherlands, April and May 2020. Euro Surveill. 2020;25:2001005. doi: 10.2807/1560-7917.ES.2020.25.23.2001005.
107. Segalés J, Puig M, Rodon J, Avila-Nieto C, Carrillo J, Cantero G et al. Detection of SARS-CoV-2 in a cat owned by a COVID-19-affected patient in Spain. PNAS. 2020;117(40):24790-24793. doi: 10.1073/pnas.2010817117
108. Hughes J, Allen RC, Baguelin M, Hampson K, Baillie GJ, Elton D et al. Transmission of equine influenza virus during an outbreak is characterized by frequent mixed infections and loose transmission bottlenecks. PLoS Pathog. 2012;8. doi:10.1371/journal.ppat.1003081.
109. Worby CJ, Lipsitch M, Hanage WP. Shared genomic variants: identification of transmission routes using pathogen deep-sequence data. Am J Epidemiol. 2017;186:1209-16. doi: 10.1093/aje/kwx182.
110. Cotten M, Lam TT, Watson SJ, Palser AL, Petrova V, Grant P et al. Full-genome deep sequencing and phylogenetic analysis of novel human betacoronavirus. Emerg Infect Dis. 2013;19:736-42B. doi: 10.3201/eid1905.130057.
111. Shen Z, Xiao Y, Kang L, Ma W, Shi L, Zhang L et al. Genomic diversity of SARS-CoV-2 in coronavirus disease 2019 patients. Clin Infect Dis. 2020; 71(15):713-720 doi: 10.1093/cid/ciaa203.
112. Grubaugh ND, Gangavarapu K, Quick J, Matteson NL, De Jesus JG, Main BJ et al. An amplicon-based sequencing framework for accurately measuring intrahost virus diversity using primalseq and ivar. Genome Biol. 2019;20:8. doi: 10.1186/s13059-018-1618-7.
113. Volz E, Baguelin M, Bhatia S, Boonyasiri A, Cori A, Cucunubá Z et al. Report 5 –phylogenetic analysis of SARS-CoV-2. London: Imperial College; 2020 (disease-epidemiology/mrc-global-infectious-disease-analysis/covid-19/report-5-phylogenetics-of-sars-cov-2/, accessed 26 June 2020).
114. Stadler T, Kühnert D, Bonhoeffer S, Drummond AJ. Birth–death skyline plot reveals temporal changes of epidemic spread in HIV and hepatitis C virus (HCV). Proc Natl Acad Sci USA. 2013;110:228-33. doi: 10.1073/pnas.1207965110.
115. Boskova V, Bonhoeffer S, Stadler T. Inference of epidemiological dynamics based on simulated phylogenies using birth-death and coalescent models. PLoS Comput Biol. 2014;10:e1003913. doi: 10.1371/journal.pcbi.1003913.
116. Volz EM, Frost SDW. Sampling through time and phylodynamic inference with coalescent and birth–death models. J R Soc Interface. 2014;11. doi: 10.1098/rsif.2014.0945.
117. Li LM, Grassly NC, Fraser C. Quantifying transmission heterogeneity using both pathogen phylogenies and incidence time series. Mol Biol Evol. 2017;34:2982-95. doi:10.1093/molbev/msx195.
118. Koelle K, Rasmussen DA. Rates of coalescence for common epidemiological models at equilibrium. J R Soc Interface. 2012;9:997-1007. doi: 10.1098/rsif.2011.0495.
119. Vaughan TG, Leventhal GE, Rasmussen DA, Drummond AJ, Welch D, Stadler T. Estimating epidemic incidence and prevalence from genomic data. Mol Biol Evol. 2019;36:1804-16. doi: 10.1093/molbev/msz106.
120. Volz EM, Siveroni I. Bayesian phylodynamic inference with complex models. PLoS Comput Biol. 2018;14:e1006546. doi: 10.1371/journal.pcbi.1006546.
121. Laboratory biosafety guidance related to coronavirus disease (COVID-19). Geneva: World Health Organization; 2020 (https://apps.who.int/iris/handle/10665/332076, accessed 21 November 2020).
122. Wang W, Xu Y, Gao R, Lu R, Han K, Wu G et al. Detection of SARS-CoV-2 in different types of clinical specimens. JAMA. 2020;323:1843-1844. doi: 10.1001/jama.2020.3786.
123. Chen W, Lan Y, Yuan X, Deng X, Li Y, Cai X et al. Detectable 2019-nCoV viral RNA in blood is a strong indicator for the further clinical severity. Emerg Microbes Infect. 2020;9:469-73. doi: 10.1080/22221751.2020.1732837.
124. Chen X, Zhao B, Qu Y, Chen Y, Xiong J, Feng Y et al. Detectable serum SARS-CoV-2 viral load (RNAaemia) is closely correlated with drastically elevated interleukin 6 (il-6) level in critically ill COVID-19 patients. Clin Infect Dis. 2020; 71(8):1937-1942. doi:10.1093/cid/ciaa449.
125. Corman VM, Rabenau HF, Adams O, Oberle D, Funk MB, Keller-Stanislawski B et al. SARS-CoV-2 asymptomatic and symptomatic patients and risk for transfusion transmission. Transfusion. 2020; 60(6):1119-1122 doi: 10.1111/trf.15841.
126. Zhang W, Du RH, Li B, Zheng XS, Yang XL, Hu B et al. Molecular and serological investigation of 2019-nCoV infected patients: implication of multiple shedding routes. Emerg Microbes Infect. 2020;9:386-9. doi: 10.1080/22221751.2020.1729071.
127. Winichakoon P, Chaiwarith R, Liwsrisakun C, Salee P, Goonna A, Limsukon A et al. Negative nasopharyngeal and oropharyngeal swabs do not rule out COVID-19. J Clin Microbiol. 2020;58. doi: 10.1128/JCM.00297-20.
128. Ek P, Bottiger B, Dahlman D, Hansen KB, Nyman M, Nilsson AC. A combination of naso and oropharyngeal swabs improves the diagnostic yield of respiratory viruses in adult emergency department patients. Infect Dis (Lond). 2019;51:241-8. doi:10.1080/23744235.2018.1546055.
129. Hammitt LL, Kazungu S, Welch S, Bett A, Onyango CO, Gunson RN et al. Added value of an oropharyngeal swab in detection of viruses in children hospitalized with lower respiratory tract infection. J Clin Microbiol. 2011;49:2318-20. doi: 10.1128/JCM.02605-10 130. The COVID-19 Investigation Team. Clinical and virologic characteristics of the first 12 patients with coronavirus disease 2019 (COVID-19) in the United States. Nat Med. 2020;26:861-868. doi: 10.1038/s41591-020-0877-5.
131. Sutjipto HL, Yant TJ, Mendis SM, Abdad MY, Marimuthu K, Ng OT et al. The effect of sample site, illness duration and the presence of pneumonia on the detection of SARS-CoV-2 by real-time reverse-transcription pcr. Open Forum Infect Dis. 2020; 7(9):ofaa335. doi: 10.1093/ofid/ofaa335.
132. Zou L, Ruan F, Huang M, Liang L, Huang H, Hong Z et al. SARS-CoV-2 viral load in upper respiratory specimens of infected patients. N Engl J Med. 2020;382:1177-9. doi: 10.1056/NEJMc2001737.
133. Lai CKC, Chen Z, Lui G, Ling L, Li T, Wong MCS et al. Prospective study comparing deep-throat saliva with other respiratory tract specimens in the diagnosis of novel coronavirus disease (COVID-19). J Infect Dis. 2020; 222(10):1612-1619. doi:10.1093/infdis/jiaa487.
134. Liu R, Han H, Liu F, Lv Z, Wu K, Liu Y et al. Positive rate of RT-PCR detection of SARS-CoV-2 infection in 4880 cases from one hospital in Wuhan, China, from Jan to Feb 2020. Clin Chim Acta. 2020;505:172-5. doi: 10.1016/j.cca.2020.03.009.
135. Huang Y, Chen S, Yang Z, Guan W, Liu D, Lin Z et al. SARS-CoV-2 viral load in clinical samples from critically ill patients. Am J Respir Crit Care Med. 2020;201:1435-8. doi: 10.1164/rccm.202003-0572LE.
136. Williams E, Bond K, Zhang B, Putland M, Williamson DA. Saliva as a non-invasive specimen for detection of SARS-CoV-2. J Clin Microbiol. 2020; 24(5):422-427. doi: 10.1128/JCM.00776-20. 68
137. Pasomsub E, Watcharananan SP, Boonyawat K, Janchompoo P, Wongtabtim G, Suksuwan W et al. Saliva sample as a non-invasive specimen for the diagnosis of coronavirus disease-2019 (COVID-19): a cross-sectional study. Clin Microbiol Infect. 2020. doi: 10.1016/j.cmi.2020.05.001.
138. Yang JR, Deng DT, Wu N, Yang B, Li HJ, Pan XB. Persistent viral RNA positivity during the recovery period of a patient with SARS-CoV-2 infection. J Med Virol. 2020; 92(9):1681-1683. doi: 10.1002/jmv.25940.
139. Guo WL, Jiang Q, Ye F, Li SQ, Hong C, Chen LY et al. Effect of throat washings on detection of 2019 novel coronavirus. Clin Infect Dis. 2020; 71(8):1980-1981. doi: 10.1093/cid/ciaa416.
140. To KK, Tsang OT, Leung WS, Tam AR, Wu TC, Lung DC et al. Temporal profiles of viral load in posterior oropharyngeal saliva samples and serum antibody responses during infection by SARS-CoV-2: an observational cohort study. Lancet Infect Dis. 2020;20:565-74. doi: 10.1016/S1473-3099(20)30196-1.
141. Azzi L, Carcano G, Gianfagna F, Grossi P, Gasperina D, Genoni A et al. Saliva is a reliable tool to detect SARS-CoV-2. J Infect. 2020;81. doi: 10.1016/j.jinf.2020.04.005.
142. McCormick-Baw C, Morgan K, Gaffney D, Cazares Y, Jaworski K, Byrd A et al. Saliva as an alternate specimen source for detection of SARS.CoV-2 in symptomatic patients using cepheid xpert xpress SARS-CoV-2. J Clin Microbiol. 2020. doi:10.1128/JCM.01109-20.
143. Wyllie AL, Fournier J, Casanovas-Massana A, Campbell M, Tokuyama M, Vijayakumar P et al. Saliva or nasopharyngeal swab specimens for detection of SARS-CoV-2. 383(13):1283-1286. N Engl J Med. 2020. doi: 10.1056/NEJMc2016359.
144. Lescure FX, Bouadma L, Nguyen D, Parisey M, Wicky PH, Behillil S et al. Clinical and virological data of the first cases of COVID-19 in Europe: a case series. Lancet Infect Dis. 2020; 20(6):697-706. doi: 10.1016/S1473-3099(20)30200-0.
145. Xing YH, Ni W, Wu Q, Li WJ, Li GJ, Wang WD et al. Prolonged viral shedding in feces of pediatric patients with coronavirus disease 2019. J Microbiol Immunol Infect. 2020; 53(3):473-480. doi: 10.1016/j.jmii.2020.03.021.
146. Zheng S, Fan J, Yu F, Feng B, Lou B, Zou Q et al. Viral load dynamics and disease severity in patients infected with SARS-CoV-2 in Zhejiang province, China, January-March 2020: retrospective cohort study. BMJ. 2020;369:1443. doi: 10.1136/bmj.m1443.
147. Wong MC, Huang J, Lai C, Ng R, Chan FKL, Chan PKS. Detection of SARS-CoV-2 RNA in fecal specimens of patients with confirmed COVID-19: a meta-analysis. J Infect. 2020;81:e31-e8. doi: 10.1016/j.jinf.2020.06.012.
148. Tang JW, To KF, Lo AW, Sung JJ, Ng HK, Chan PK. Quantitative temporal-spatial distribution of severe acute respiratory syndrome-associated coronavirus (SARS-CoV) in post-mortem tissues. J Med Virol. 2007;79:1245-53. doi: 10.1002/jmv.20873.
149. Nicholls JM, Poon LL, Lee KC, Ng WF, Lai ST, Leung CY et al. Lung pathology of fatal severe acute respiratory syndrome. Lancet. 2003;361:1773-8. doi: 10.1016/s0140-6736(03)13413-7.
150. Pomara C, Li Volti G, Cappello F. COVID-19 deaths: are we sure it is pneumonia? Please, autopsy, autopsy, autopsy! J Clin Med. 2020;9. doi: 10.3390/jcm9051259.
151. Salerno M, Sessa F, Piscopo A, Montana A, Torrisi M, Patane F et al. No autopsies on COVID-19 deaths: a missed opportunity and the lockdown of science. J Clin Med. 2020;9. doi: 10.3390/jcm9051472.
152. Hanley B, Lucas SB, Youd E, Swift B, Osborn M. Autopsy in suspected COVID-19 cases. J Clin Pathol. 2020;73:239-42. doi: 10.1136/jclinpath-2020-206522.
153. Basso C, Calabrese F, Sbaraglia M, Del Vecchio C, Carretta G, Saieva A et al. Feasibility of postmortem examination in the era of COVID-19 pandemic: the experience of a northeast Italy university hospital. Virchows Arch. 2020 477(3):341-347. doi:10.1007/s00428-020-02861-1.
154. Tian S, Xiong Y, Liu H, Niu L, Guo J, Liao M et al. Pathological study of the 2019 novel coronavirus disease (COVID-19) through postmortem core biopsies. Mod Pathol. 2020;33:1007-14. doi: 10.1038/s41379-020-0536-x.
155. Sekulic M, Harper H, Nezami BG, Shen DL, Sekulic SP, Koeth AT et al. Molecular detection of SARS-CoV-2 infection in FFPE samples and histopathologic findings in fatal SARS-CoV-2 cases. Am J Clin Pathol. 2020; 154(2):190-200. doi:10.1093/ajcp/aqaa091.
156. Park WB, Kwon NJ, Choi SJ, Kang CK, Choe PG, Kim JY et al. Virus isolation from the first patient with SARS-CoV-2 in Korea. J Korean Med Sci. 2020;35:e84. doi: 10.3346/jkms.2020.35.e84.
157. Le TQM, Takemura T, Moi ML, Nabeshima T, Nguyen LKH, Hoang VMP et al. Severe acute respiratory syndrome coronavirus 2 shedding by travelers, Vietnam, 2020. Emerg Infect Dis . 2020;26:1624-6. doi: 10.3201/eid2607.200591.
158. Pan Y, Zhang D, Yang P, Poon LLM, Wang Q. Viral load of SARS-CoV-2 in clinical samples. Lancet Infect Dis. 2020;20:411-2. doi: 10.1016/S1473-3099(20)30113-4.
159. Wyllie AL, Fournier J, Casanovas-Massana A, Campbell M, Tokuyama M, Vijayakumar P et al. Saliva or nasopharyngeal swab specimens for detection of SARS-CoV-2. N Eng J Med. 2020;383:1283-6. doi: 10.1101/2020.04.16.20067835.
160. Wolfel R, Corman VM, Guggemos W, Seilmaier M, Zange S, Muller MA et al. Virological assessment of hospitalized patients with COVID-2019. Nature. 2020; 581(7809):465-469. doi: 10.1038/s41586-020-2196-x.
161. MacCannell D. SARS-CoV-2 sequencing. 2020 (CoV-2_Sequencing, accessed 1 November 2020).
162. Quince C, Walker AW, Simpson JT, Loman NJ, Segata N. Shotgun metagenomics, from sampling to analysis. Nat Biotechnol. 2017;35:833-44. doi: 10.1038/nbt.3935.
163. Bragg L, Tyson GW. Metagenomics using next-generation sequencing. Methods in Mol Biol. 2014;1096:183-201. doi: 10.1007/978-1-62703-712-9_15.
164. Xiao M, Liu X, Ji J, Li M, Li J, Yang L et al. Multiple approaches for massively parallel sequencing of SARS-CoV-2 genomes directly from clinical samples. Genome Med. 2020;12:57. doi: 10.1186/s13073-020-00751-4.
166. Vogels CBF, Brito AF, Wyllie AL, Fauver JR, Ott IM, Kalinich CC et al. Analytical sensitivity and efficiency comparisons of SARS-CoV-2 RT–qPCR primer–probe sets. Nat Microbiol. 2020:1-7. doi: 10.1038/s41564-020-0761-6.
167. Quick J, Grubaugh ND, Pullan ST, Claro IM, Smith AD, Gangavarapu K et al. Multiplex PCR method for Minion and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nature Protoc. 2017;12:1261-76. doi:10.1038/nprot.2017.066.
168. Matteson N. Primalseq: generation of tiled virus amplicons for miseq sequencing. Protocolsio. 2020. doi: 10.17504/protocols.io.bez7jf9n.
169. Gordon P, Mabon P. Nanostripper2020 (https://github.com/nodrogluap/nanostripper, accessed 15 July 2020).
170. Wood DE, Lu J, Langmead B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019;20:257. doi: 10.1186/s13059-019-1891-0.
171. Ounit R, Wanamaker S, Close TJ, Lonardi S. Clark. Fast and accurate classification of metagenomic and genomic sequences using discriminative k-mers. BMC Genomics. 2015;16:236. doi: 10.1186/s12864-015-1419-2.
172. Wu TD, Reeder J, Lawrence M, Becker G, Brauer MJ. Gmap and gsnap for genomic sequence alignment: enhancements to speed, accuracy, and functionality. Methods Mol Biol. 2016;1418:283-334. doi: 10.1007/978-1-4939-3578-9_15.
174. Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnetjournal. 2011;17:10-12.
175. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114-20. doi: 10.1093/bioinformatics/btu170.
176. NCBI. Severe acute respiratory syndrome coronavirus 2 isolate Wuhan-Hu-1, complete genome. 2020 (https://www.ncbi.nlm.nih.gov/nuccore/NC_045512.2, accessed 1 November 2020).
177. Langmead B, Salzberg SL. Fast gapped-read alignment with bowtie 2. Nat Methods. 2012;9:357-9. doi: 10.1038/nmeth.1923.
178. Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34:3094-100. doi: 10.1093/bioinformatics/bty191.
179. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754-60. doi: 10.1093/bioinformatics/btp324.
180. Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011;27:2987-93. doi: 10.1093/bioinformatics/btr509.
181. Loman NJ, Quick J, Simpson JT. A complete bacterial genome assembled de novo using only nanopore sequencing data. Nat Methods. 2015;12:733-5. doi: 10.1038/nmeth.3444.
182. Li W, Jaroszewski L, Godzik A. Clustering of highly homologous sequences to reduce the size of large protein databases. Bioinformatics. 2001;17:282-3. doi: 10.1093/bioinformatics/17.3.282.
183. Hadfield J, Megill C, Bell SM, Huddleston J, Potter B, Callender C et al. Nextstrain: real-time tracking of pathogen evolution. Bioinformatics. 2018;34:4121-3. doi:10.1093/bioinformatics/bty407.
184. Hong SL, Dellicour S, Vrancken B, Suchard MA, Pyne MT, Hillyard DR et al. In search of covariates of HIV-1 subtype B spread in the United States—a cautionary tale of large-scale Bayesian phylogeography. Viruses. 2020;12:182. doi: 10.3390/v12020182.
185. Katoh K, Standley DM. Mafft multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772-80. doi: 10.1093/molbev/mst010.
187. Wymant C, Blanquart F, Golubchik T, Gall A, Bakker M, Bezemer D et al. Easy and accurate reconstruction of whole HIV genomes from short-read sequence data with shiver. Virus Evol. 2018;4. doi: 10.1093/ve/vey007.
188. Singer J, Gifford R, Cotten M, Robertson D. CoV-glue: a web application for tracking SARS-CoV-2 genomic variation. Preprints 2020; 2020060225. doi: 10.20944/preprints202006.0225.v1.
189. Rambaut A, Holmes EC, O’Toole Á, Hill V, McCrone JT, Ruis C et al. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat Microbiol. 2020:1-5. doi: 10.1038/s41564-020-0770-5.
190. Rambaut A, Lam TT, Carvalho LM, Pybus OG. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-o-gen). Virus Evol. 2016;2. doi: 10.1093/ve/vew007.
192. Martin DP, Murrell B, Golden M, Khoosal A, Muhire B. Rdp4: detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015;1. doi: 10.1093/ve/vev003.
193. Price MN, Dehal PS, Arkin AP. Fasttree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol Biol Evol. 2009;26:1641-50. doi: 10.1093/molbev/msp077.
194. Darriba D, Posada D, Kozlov AM, Stamatakis A, Morel B, Flouri T. Modeltest-ng: a new and scalable tool for the selection of DNA and protein evolutionary models. Mol Biol Evol. 2019; 37(1):291-294. doi: 10.1093/molbev/msz189.
195. Guindon S, Dufayard J-F, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010;59:307-21. doi: 10.1093/sysbio/syq010.
196. Kozlov AM, Darriba D, Flouri T, Morel B, Stamatakis A. RAxML-NG: A fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics. 2019;35:4453-5. doi: 10.1093/bioinformatics/btz305.
197. Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015;32:268-74. doi: 10.1093/molbev/msu300.
198. Minh BQ, Schmidt HA, Chernomor O, Schrempf D, Woodhams MD, von Haeseler A et al. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol Biol Evol. 2020;37:1530-4. doi: 10.1093/molbev/msaa015.
199. Suchard MA, Lemey P, Baele G, Ayres DL, Drummond AJ, Rambaut A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018;4:vey016. doi: 10.1093/ve/vey016.
200. Bouckaert R, Vaughan TG, Barido-Sottani J, Duchêne S, Fourment M, Gavryushkina A et al. BEAST 2.5: an advanced software platform for Bayesian evolutionary analysis. PLoS Comput Biol. 2019;15:e1006650. doi: 10.1371/journal.pcbi.1006650.
201. To T-H, Jung M, Lycett S, Gascuel O. Fast dating using least-squares criteria and algorithms. Syst Biol. 2016;65:82-97. doi: 10.1093/sysbio/syv068. 72
202. Kong S, Sánchez‐Pacheco SJ, Murphy RW. On the use of median-joining networks in evolutionary biology. Cladistics. 2016;32:691-9. doi: 10.1111/cla.12147.
203. Kumar S, Stecher G, Li M, Knyaz C, Tamura K. Mega x: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 2018;35:1547-9. doi: 10.1093/molbev/msy096.
204. Argimón S, Abudahab K, Goater RJE, Fedosejev A, Bhai J, Glasner C et al. Microreact: visualizing and sharing data for genomic epidemiology and phylogeography. Microb Genom. 2016;2. doi: 10.1099/mgen.0.000093.
205. Nadeau SA, Vaughan TG, Sciré J, Huisman JS, Stadler T. The origin and early spread of SARS-CoV-2 in Europe. 2020 (http://medrxiv.org/lookup/doi/10.1101/2020.06.10.20127738, accessed 17 July 2020).



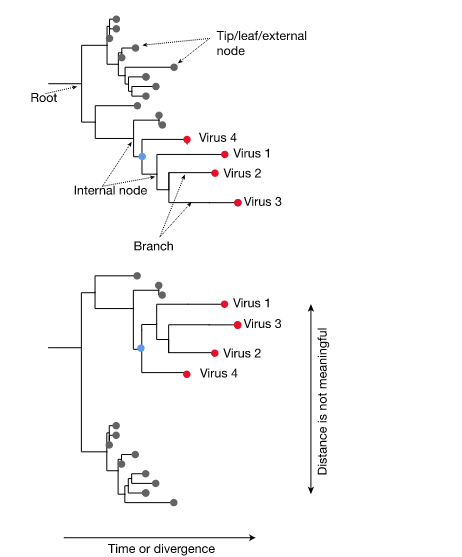


















")





