Pfizer Comirnaty non può essere utilizzato sui minori di 12 anni
Una volta che è nota la direzione che ha preso la scienza “marcia” e soprattutto le intenzioni di tutti questi personaggi “oscuri” che in questo momento stanno dominando il mondo, non è certo difficile immaginare la loro linea di pensiero e soprattutto le loro macabre intenzioni che già da più di venti anni stanno palesando.
Pfizer Comirnaty non può essere utilizzato sui minori di 12 anni
Bimba della Pennsylvania soffre di ictus ed emorragia cerebrale 7 giorni dopo essere stata….avvelenata….
Ricevo e pubblico.
“La mia bellissima e meravigliosa nipote Harper.
È così dolce.
Per favore pregate per lei nel potente nome di Yeshua per la sua guarigione.
Le è stata fatta l’iniezione 7 giorni fa, la prima nella sua vita, ed ieri sera è stata portata in terapia intensiva perché ha avuto un ictus ed un’emorragia cerebrale.
Grazie per le vostre preghiere. “
Ora potete iniziare a leggere ciò che ho pubblicato sotto, fate MOLTA attenzione alle evidenti contraddizioni all’interno delle cosiddette istruzioni (evidenziate in grassetto), e non solo a quelle…
Medicinale sottoposto a monitoraggio addizionale.
Ciò permetterà la rapida identificazione di nuove informazioni sulla sicurezza.
Agli operatori sanitari è richiesto di segnalare qualsiasi reazione avversa sospetta.
Vedere paragrafo 4.8 per informazioni sulle modalità di segnalazione delle reazioni avverse. 1.DENOMINAZIONE DEL MEDICINALE Comirnaty 30 microgrammi/dose concentrato per dispersione iniettabile. Vaccino a mRNA anti-COVID-19 (modificato a livello dei nucleosidi). 2.COMPOSIZIONE QUALITATIVA E QUANTITATIVA Flaconcino multidose da diluire prima dell’uso. Ogni flaconcino (0,45 mL) contiene 6 dosi da 0,3 mL dopo la diluizione, vedere paragrafi 4.2 e 6.6. Ogni dose (0,3 mL) contiene 30 microgrammi di tozinameran, un vaccino a mRNA anti-COVID-19 (inserito in nanoparticelle lipidiche). Tozinameran è un RNA messaggero (mRNA) a singola elica con capping in 5’, prodotto mediante trascrizione in vitro senza l’ausilio di cellule (cell-free) dai corrispondenti DNA stampo, che codifica per la proteina virale spike (S) di SARS-CoV-2. Per l’elenco completo degli eccipienti, vedere paragrafo 6.1.
3.FORMA FARMACEUTICA Concentrato per dispersione iniettabile (concentrato sterile). Il vaccino si presenta come una dispersione congelata di colore da bianco a biancastro (pH: 6,9-7,9). 4.INFORMAZIONI CLINICHE 4.1Indicazioni terapeutiche Comirnaty è indicato per l’immunizzazione attiva per la prevenzione di COVID-19, malattia causata dal virus SARS-CoV-2, in soggetti di età pari o superiore a 12 anni. L’uso di questo vaccino deve essere in accordo con le raccomandazioni ufficiali. 4.2Posologia e modo di somministrazione Posologia
Soggetti di età pari o superiore a 12 anni Comirnaty viene somministrato per via intramuscolare dopo diluizione come ciclo primario di 2 dosi (da 0,3 mL ciascuna).
Si raccomanda di somministrare la seconda dose 3 settimane dopo la prima dose (vedere paragrafi 4.4 e 5.1). È possibile somministrare una dose di richiamo (terza dose) di Comirnaty per via intramuscolare almeno 6 mesi dopo la seconda dose a soggetti di età pari o superiore a 18 anni. La decisione in merito alle tempistiche e ai destinatari della terza dose di Comirnaty deve essere presa sulla base dei dati disponibili sull’efficacia del vaccino, tenendo in considerazione la limitata disponibilità di dati relativi alla sicurezza (vedere paragrafi 4.4 e 5.1). L’intercambiabilità di Comirnaty con vaccini anti-COVID-19 di altri produttori per completare il ciclo primario di vaccinazione o la dose di richiamo (terza dose) non è stata stabilita.
I soggetti che hanno ricevuto 1 dose di Comirnaty devono ricevere una seconda dose di Comirnaty per completare il ciclo primario di vaccinazione e per eventuali dosi aggiuntive.
Le dosi di Comirnaty 30 microgrammi/dose concentrato per dispersione iniettabile dopo diluizione e Comirnaty 30 microgrammi/dose dispersione per preparazione iniettabile sono considerate intercambiabili. Soggetti severamente immunocompromessi di età pari o superiore a 12 anni È possibile somministrare una terza dose almeno 28 giorni dopo la seconda dose a soggetti severamente immunocompromessi (vedere paragrafo 4.4). Popolazione pediatrica La sicurezza e l’efficacia di Comirnaty nella popolazione pediatrica di età inferiore a 12 anni non sono state ancora stabilite. I dati disponibili sono limitati. Anziani Non è necessario alcun aggiustamento posologico nei soggetti anziani di età ≥65 anni.
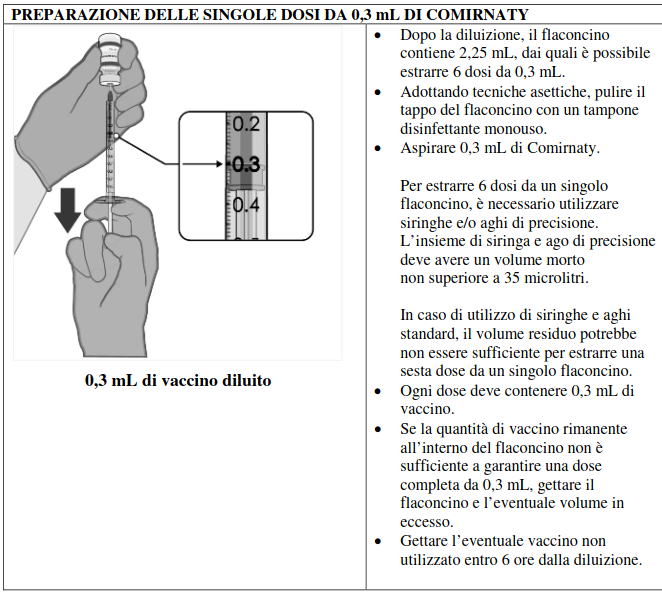
La sicurezza e l’immunogenicità di una dose di richiamo (terza dose) di Comirnaty in soggetti di età pari o superiore a 65 anni si basano sui dati relativi alla sicurezza e all’immunogenicità negli adulti di età compresa fra 18 e 55 anni. Modo di somministrazione Comirnaty deve essere somministrato per via intramuscolare dopo diluizione (vedere paragrafo 6.6). Dopo la diluizione, i flaconcini di Comirnaty contengono 6 dosi da 0,3 mL di vaccino.
Per estrarre 6 dosi da un singolo flaconcino, è necessario utilizzare siringhe e/o aghi di precisione.
L’insieme di siringa e ago di precisione deve avere un volume morto non superiore a 35 microlitri.
In caso di utilizzo di siringhe e aghi standard, il volume residuo potrebbe non essere sufficiente per estrarre una sesta dose da un singolo flaconcino.
Indipendentemente dal tipo di siringa e di ago : •ogni dose deve contenere 0,3 mL di vaccino; •se la quantità di vaccino rimanente all’interno del flaconcino non è sufficiente a garantire una dose completa da 0,3 mL, gettare il flaconcino e l’eventuale volume in eccesso; •non mescolare residui di vaccino provenienti da flaconcini diversi. La sede preferita è la regione deltoidea del braccio. Il vaccino non deve essere iniettato per via endovenosa, sottocutanea o intradermica. Il vaccino non deve essere miscelato con altri vaccini o medicinali nella stessa siringa. Per le precauzioni da adottare prima della somministrazione del vaccino, vedere paragrafo 4.4.
Per le istruzioni relative allo scongelamento, alla manipolazione e allo smaltimento del vaccino, vedere paragrafo 6.6. 4.3Controindicazioni Ipersensibilità al principio attivo o ad uno qualsiasi degli eccipienti elencati al paragrafo 6.1. 4.4Possibili effetti indesiderati
Tracciabilità Al fine di migliorare la tracciabilità dei medicinali biologici, il nome e il numero di lotto del medicinale somministrato devono essere chiaramente registrati. Raccomandazioni generali
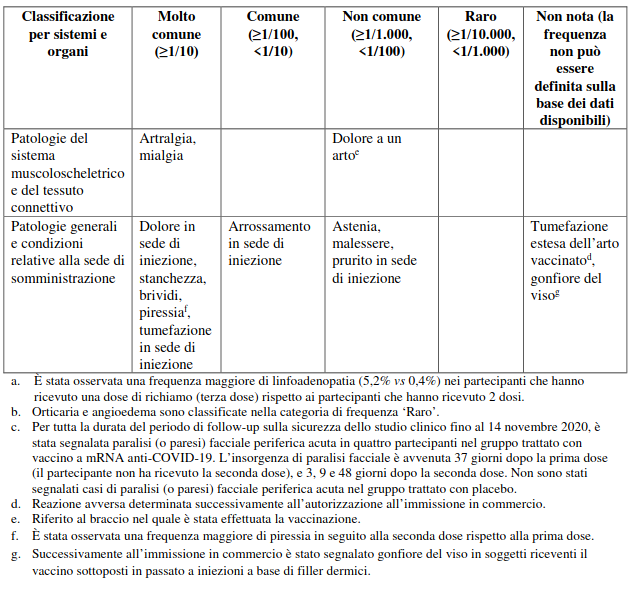
Ipersensibilità e anafilassi Sono stati segnalati casi di anafilassi. Devono essere sempre immediatamente disponibili trattamento e assistenza medici adeguati nel caso di comparsa di una reazione anafilattica in seguito alla somministrazione del vaccino. Dopo la vaccinazione si raccomanda un attento monitoraggio per almeno 15 minuti. Non somministrare la seconda dose del vaccino a soggetti che abbiano manifestato anafilassi alla prima dose di Comirnaty. Miocardite e pericardite Dopo la vaccinazione con Comirnaty sono stati osservati casi molto rari di miocardite e pericardite, verificatisi principalmente nei 14 giorni successivi alla vaccinazione, più spesso dopo la seconda dose e nei giovani di sesso maschile. I dati a disposizione suggeriscono che il decorso della miocardite e della pericardite dopo la vaccinazione non è diverso da quello della miocardite o della pericardite in generale. Gli operatori sanitari devono prestare attenzione ai segni e ai sintomi di miocardite e pericardite. Le persone vaccinate devono essere istruite a rivolgersi immediatamente al medico qualora dopo la vaccinazione sviluppino sintomi indicativi di miocardite o pericardite, quali dolore toracico (acuto e persistente), respiro affannoso o palpitazioni. Gli operatori sanitari devono consultare le linee guida e/o specialisti per diagnosticare e trattare tale affezione. Il rischio di miocardite in seguito a una terza dose di Comirnaty non è ancora stato caratterizzato. Reazioni correlate all’ansia In associazione alla procedura di vaccinazione stessa possono verificarsi reazioni correlate all’ansia, incluse reazioni vasovagali (sincope), iperventilazione o reazioni correlate allo stress (ad es. capogiro, palpitazioni, aumenti della frequenza cardiaca, alterazioni della pressione arteriosa, sensazioni di formicolio, sudorazione).
Le reazioni correlate allo stress sono temporanee e si risolvono spontaneamente.
Ai soggetti deve essere raccomandato di segnalare eventuali sintomi all’operatore addetto alla vaccinazione, perché possa valutarli.
È importante che vengano adottate precauzioni per evitare lesioni da svenimento. Malattia concomitante La vaccinazione deve essere rimandata nei soggetti affetti da uno stato febbrile acuto severo o da un’infezione acuta.
La presenza di un’infezione lieve e/o di febbre di lieve entità non deve comportare il rinvio della vaccinazione. Trombocitopenia e disturbi della coagulazione Come per tutte le iniezioni intramuscolari, il vaccino deve essere somministrato con cautela nei soggetti sottoposti a terapia anticoagulante oppure affetti da trombocitopenia o qualsiasi disturbo della coagulazione (ad es. emofilia), poiché in questi soggetti possono verificarsi sanguinamenti o lividi a seguito di una somministrazione intramuscolare.
Soggetti immunocompromessi L’efficacia e la sicurezza del vaccino non sono state valutate nei soggetti immunocompromessi, compresi quelli in terapia immunosoppressiva. L’efficacia di Comirnaty potrebbe essere inferiore nei soggetti immunocompromessi. La raccomandazione di considerare una terza dose in soggetti severamente immunocompromessi si basa su un’evidenza sierologica limitata ricavata da una serie di casi in letteratura sulla gestione clinica di pazienti con immunocompromissione iatrogena in seguito a trapianto di organo solido (vedere paragrafo 4.2). Durata della protezione La durata della protezione offerta dal vaccino non è nota ; sono tuttora in corso studi clinici volti a stabilirla. Limitazioni dell’efficacia del vaccino Come per tutti i vaccini, la vaccinazione con Comirnaty potrebbe non proteggere tutti coloro che lo ricevono.
I soggetti potrebbero non essere completamente protetti fino a 7 giorni dopo la seconda dose del vaccino. Eccipienti Questo vaccino contiene potassio, meno di 1 mmol (39 mg) per dose, cioè è essenzialmente ‘senza potassio’. Questo vaccino contiene meno di 1 mmol (23 mg) di sodio per dose, cioè è essenzialmente ‘senza sodio’. 4.5Interazioni con altri medicinali ed altre forme d’interazione Non sono stati effettuati studi d’interazione. La somministrazione concomitante di Comirnaty con altri vaccini non è stata studiata. 4.6Fertilità, gravidanza e allattamento Gravidanza I dati relativi all’uso di Comirnaty in donne in gravidanza sono in numero limitato. Gli studi sugli animali non indicano effetti dannosi diretti o indiretti su gravidanza, sviluppo embrionale/fetale, parto o sviluppo post-natale (vedere paragrafo 5.3). La somministrazione di Comirnaty durante la gravidanza deve essere presa in considerazione solo se i potenziali benefici sono superiori ai potenziali rischi per la madre e per il feto. Allattamento Non è noto se Comirnaty sia escreto nel latte materno. Fertilità Gli studi sugli animali non indicano effetti dannosi diretti o indiretti di tossicità riproduttiva (vedere paragrafo 5.3). 4.7Effetti sulla capacità di guidare veicoli e sull’uso di macchinari Comirnaty non altera o altera in modo trascurabile la capacità di guidare veicoli e di usare macchinari. Tuttavia, alcuni degli effetti menzionati al paragrafo 4.8 possono influenzare temporaneamente la capacità di guidare veicoli o usare macchinari. 4.8Effetti indesiderati Riassunto del profilo di sicurezza La sicurezza di Comirnaty è stata valutata in soggetti di età pari o superiore a 12 anni nel corso di 2 studi clinici che hanno coinvolto 23.205 partecipanti (di cui 22.074 di età pari o superiore a 16 anni e 1.131 adolescenti di età compresa fra 12 e 15 anni) i quali hanno ricevuto almeno una dose di Comirnaty. Il profilo di sicurezza complessivo di Comirnaty negli adolescenti di età compresa fra 12 e 15 anni si è dimostrato simile a quello osservato nei partecipanti di età pari o superiore a 16 anni. Inoltre, 306 soggetti già arruolati, di età compresa fra 18 e 55 anni, partecipanti alla fase 3, hanno ricevuto una dose di richiamo (terza dose) di Comirnaty circa 6 mesi dopo la somministrazione della seconda dose. Il profilo di sicurezza complessivo della dose di richiamo (terza dose) si è dimostrato simile a quello osservato dopo 2 dosi. Soggetti di età pari o superiore a 16 anni – dopo 2 dosi Nello Studio 2, un totale di 22.026 partecipanti di età pari o superiore a 16 anni ha ricevuto almeno 1 dose di Comirnaty, mentre un totale di 22.021 partecipanti di età pari o superiore a 16 anni ha ricevuto placebo (compresi 138 e 145 adolescenti di 16 e 17 anni di età, rispettivamente nel gruppo trattato con vaccino e nel gruppo trattato con placebo).
Un totale di 20.519 partecipanti di età pari o superiore a 16 anni ha ricevuto 2 dosi di Comirnaty. Al momento dell’analisi dello Studio 2, con la data limite del 13 marzo 2021 per il periodo di follow-up in cieco controllato verso placebo fino alle date di apertura del cieco per i partecipanti, un totale di 25.651 (58,2%) partecipanti (13.031 trattati con Comirnaty e 12.620 trattati con placebo) di età pari o superiore a 16 anni, è stato seguito per ≥4 mesi dopo la seconda dose.
Erano inclusi un totale di 15.111 partecipanti (7.704 trattati con Comirnaty e 7.407 trattati con placebo) di età compresa fra 16 e 55 anni, e un totale di 10.540 partecipanti (5.327 trattati con Comirnaty e 5.213 trattati con placebo) di età pari o superiore a 56 anni. Le reazioni avverse più frequenti nei soggetti di età pari o superiore a 16 anni che avevano ricevuto 2 dosi sono state dolore in sede di iniezione (>80%), stanchezza (>60%), cefalea (>50%), mialgia (>40%), brividi (>30%), artralgia (>20%), piressia e tumefazione in sede di iniezione (>10%). Tali reazioni sono state generalmente di intensità da lieve a moderata e si sono risolte entro pochi giorni dalla vaccinazione.
Una frequenza leggermente inferiore di reazioni di reattogenicità è stata associata ad un’età maggiore. Il profilo di sicurezza in 545 soggetti di età pari o superiore a 16 anni che hanno ricevuto Comirnaty, risultati positivi al SARS-CoV-2 al basale, si è dimostrato simile a quello osservato nella popolazione generale. Adolescenti di età compresa fra 12 e 15 anni – dopo 2 dosi In un’analisi dello Studio 2, basata sui dati raccolti fino alla data limite del 13 marzo 2021, 2.260 adolescenti (1.131 trattati con Comirnaty e 1.129 trattati con placebo) avevano un’età compresa fra 12 e 15 anni.
Di questi, 1.308 adolescenti (660 trattati con Comirnaty e 648 trattati con placebo) sono stati seguiti per almeno 2 mesi dopo la somministrazione della seconda dose di Comirnaty.
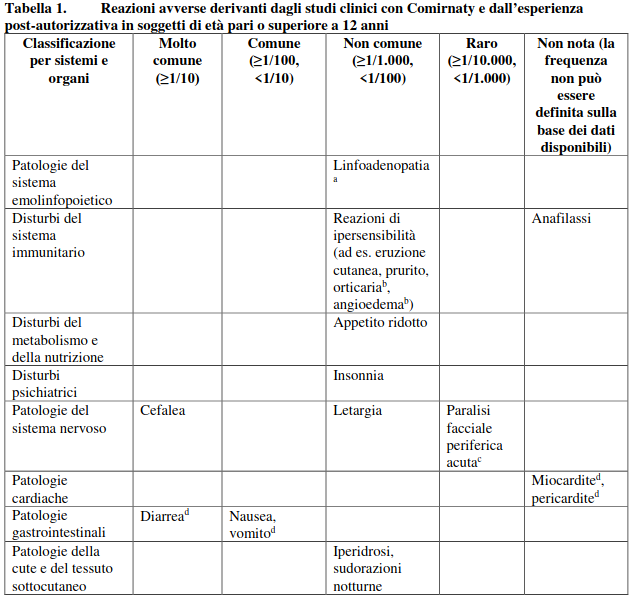
La valutazione della sicurezza dello Studio 2 è tuttora in corso. Le reazioni avverse più frequenti negli adolescenti di età compresa fra 12 e 15 anni che avevano ricevuto 2 dosi sono state dolore in sede di iniezione (>90%), stanchezza e cefalea (>70%), mialgia e brividi (>40%), artralgia e piressia (>20%). Partecipanti di età pari o superiore a 18 anni – dopo la dose di richiamo (terza dose) Dei partecipanti alla fase 2/3 dello Studio 2, un sottogruppo di 306 adulti di età compresa fra 18 e 55 anni, che avevano completato il ciclo originale di 2 dosi di Comirnaty, ha ricevuto una dose di richiamo (terza dose) di Comirnaty circa 6 mesi (intervallo: 4,8-8,0 mesi) dopo la somministrazione della seconda dose. Le reazioni avverse più frequenti nei partecipanti di età compresa fra 18 e 55 anni sono state dolore in sede di iniezione (>80%), stanchezza (>60%), cefalea (>40%), mialgia (>30%), brividi e artralgia (>20%). Tabella delle reazioni avverse derivanti dagli studi clinici e dall’esperienza post-autorizzativa in soggetti di età pari o superiore a 12 anni Le reazioni avverse osservate nel corso degli studi clinici sono elencate sotto, in base alle seguenti categorie di frequenza: molto comune (≥1/10), comune (≥1/100, <1/10), non comune (≥1/1.000, <1/100), raro (≥1/10.000, <1/1.000), molto raro (<1/10.000), non nota (la frequenza non può essere definita sulla base dei dati disponibili).
Segnalazione delle reazioni avverse sospette La segnalazione delle reazioni avverse sospette che si verificano dopo l’autorizzazione del medicinale è importante, in quanto permette un monitoraggio continuo del rapporto beneficio/rischio del medicinale.
Agli operatori sanitari è richiesto di segnalare qualsiasi reazione avversa sospetta tramite il sistema nazionale di segnalazione riportato nell’allegato V, includendo il numero di lotto, se disponibile. 4.9Sovradosaggio I dati relativi al sovradosaggio sono stati ricavati da 52 partecipanti inclusi nello studio clinico che avevano ricevuto 58 microgrammi di Comirnaty a causa di un errore di diluizione. Nei soggetti vaccinati non è stato osservato alcun incremento della reattogenicità o delle reazioni avverse. In caso di sovradosaggio, si raccomanda il monitoraggio delle funzioni vitali e l’eventuale trattamento sintomatico.
5.PROPRIETÀ FARMACOLOGICHE 5.1Proprietà farmacodinamiche Categoria farmacoterapeutica: vaccini, altri vaccini virali, codice ATC: J07BX03 Meccanismo d’azione L’RNA messaggero modificato a livello dei nucleosidi presente in Comirnaty (tozinameran) è formulato in nanoparticelle lipidiche, per consentire il rilascio dell’RNA non replicante all’interno delle cellule ospiti e dirigere l’espressione transitoria dell’antigene S di SARS-CoV-2.
L’mRNA codifica per una proteina S intera ancorata alla membrana, con due mutazioni puntiformi a livello dell’elica centrale. La mutazione di questi due aminoacidi in prolina stabilizza la proteina S in conformazione di prefusione, antigenicamente preferenziale.
Il vaccino induce sia una risposta anticorpale neutralizzante che una risposta immunitaria cellulo-mediata verso l’antigene delle proteine spike (S), che possono contribuire a proteggere contro COVID-19. Efficacia Lo Studio 2 è uno studio multicentrico, multinazionale, randomizzato, controllato verso placebo, in cieco per l’osservatore, di fase 1/2/3 per la determinazione della dose, la selezione di un potenziale vaccino e la valutazione dell’efficacia, condotto su soggetti di età pari o superiore a 12 anni.
La randomizzazione è stata stratificata per fasce d’età: da 12 a 15 anni, da 16 a 55 anni, o da 56 anni in poi, con almeno il 40% dei partecipanti nella fascia d’età ≥56 anni.
Dallo studio sono stati esclusi i soggetti immunocompromessi e quelli con pregressa diagnosi clinica o microbiologica di COVID-19.
Sono stati inclusi i soggetti con malattia stabile preesistente (definita come malattia che non avesse richiesto una modifica sostanziale della terapia né il ricovero in ospedale a causa di un peggioramento della malattia nelle 6 settimane precedenti l’arruolamento), e quelli con infezione nota e stabile da virus dell’immunodeficienza umana (HIV), da virus dell’epatite C (HCV) o da virus dell’epatite B (HBV). Efficacia nei partecipanti di età pari o superiore a 16 anni – dopo 2 dosi Nella parte di fase 2/3 dello Studio 2, sulla base dei dati raccolti fino al 14 novembre 2020, circa 44.000 partecipanti sono stati randomizzati in numero uguale a ricevere 2 dosi di vaccino a mRNA anti-COVID-19 oppure placebo.
Nelle analisi di efficacia sono stati inclusi i partecipanti che avevano ricevuto la seconda vaccinazione a distanza di 19-42 giorni dalla prima.
La maggior parte (93,1%) di coloro che avevano ricevuto il vaccino ha ricevuto la seconda dose da 19 giorni a 23 giorni dopo la dose 1.
È previsto il follow-up dei partecipanti fino a 24 mesi dopo la dose 2, per valutare la sicurezza e l’efficacia contro COVID-19.
Nello studio clinico, i partecipanti hanno dovuto osservare un intervallo minimo di 14 giorni prima e dopo la somministrazione di un vaccino antinfluenzale per poter ricevere placebo oppure vaccino a mRNA anti-COVID-19.
Nello studio clinico, i partecipanti hanno dovuto osservare un intervallo minimo di 60 giorni prima o dopo la somministrazione di emocomponenti/plasmaderivati o immunoglobuline, per tutta la durata dello studio e fino a conclusione dello stesso, per poter ricevere placebo oppure vaccino a mRNA anti-COVID-19. La popolazione per l’analisi dell’endpoint primario di efficacia era composta da 36.621 partecipanti di età pari o superiore a 12 anni (18.242 nel gruppo trattato con vaccino a mRNA anti-COVID-19 e 18.379 nel gruppo trattato con placebo) che non avevano presentato evidenza di infezione pregressa da SARS-CoV-2 fino ai 7 giorni successivi alla somministrazione della seconda dose.
Inoltre, 134 partecipanti erano di età compresa fra 16 e 17 anni (66 nel gruppo trattato con vaccino a mRNA anti-COVID-19 e 68 nel gruppo trattato con placebo), e 1.616 partecipanti erano di età ≥75 anni (804 nel gruppo trattato con vaccino a mRNA anti-COVID-19 e 812 nel gruppo trattato con placebo).
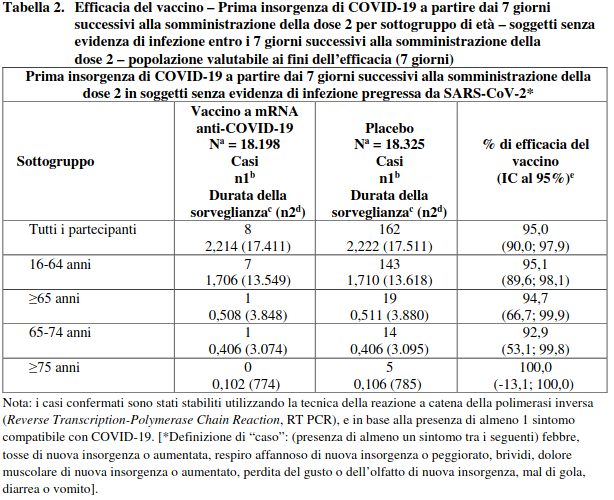
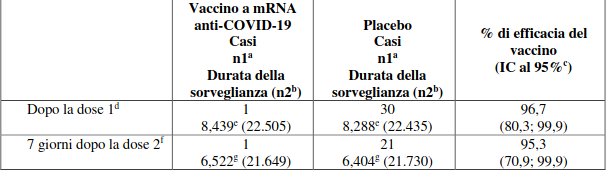
Al momento dell’analisi di efficacia primaria, i partecipanti erano stati seguiti per monitorare l’insorgenza di COVID-19 sintomatica per 2.214 persone/anno in totale nel gruppo trattato con vaccino a mRNA anti-COVID-19, e per 2.222 persone/anno in totale nel gruppo trattato con placebo. Non sono state rilevate differenze cliniche significative in termini di efficacia complessiva del vaccino nei partecipanti a rischio di COVID-19 severa, compresi quelli con 1 o più comorbilità suscettibili di aumentare il rischio di COVID-19 severa (ad es. asma, indice di massa corporea (IMC) ≥30 kg/m2, malattia polmonare cronica, diabete mellito, ipertensione). Le informazioni sull’efficacia del vaccino sono presentate nella Tabella 2. Tabella 2. Efficacia del vaccino – Prima insorgenza di COVID-19 a partire dai 7 giorni successivi alla somministrazione della dose 2 per sottogruppo di età – soggetti senza evidenza di infezione entro i 7 giorni successivi alla somministrazione della dose 2 – popolazione valutabile ai fini dell’efficacia (7 giorni)
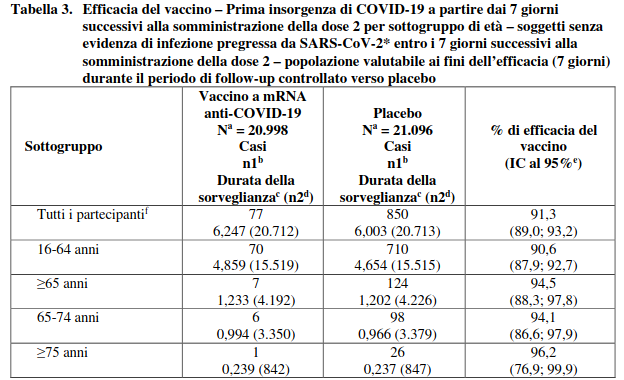
L’efficacia del vaccino a mRNA anti-COVID-19 nella prevenzione della prima insorgenza di COVID-19 a partire da 7 giorni dopo la somministrazione della seconda dose, rispetto al placebo, è risultata pari al 94,6% (intervallo di confidenza al 95% compreso fra 89,6% e 97,6%) nei soggetti di età ≥16 anni con o senza evidenza di infezione pregressa da SARS-CoV-2. Inoltre, le analisi per sottogruppi dell’endpoint primario di efficacia hanno dimostrato stime puntuali di efficacia paragonabili fra sessi, etnie e fra partecipanti con comorbilità associate a un rischio elevato di COVID-19 severa. Sono state condotte analisi di efficacia aggiornate con ulteriori casi confermati di COVID-19 raccolti durante il follow-up in cieco controllato verso placebo, equivalenti a un massimo di 6 mesi dopo la somministrazione della seconda dose nella popolazione valutabile ai fini dell’efficacia. Le informazioni aggiornate sull’efficacia del vaccino sono riportate nella Tabella 3. Tabella 3. Efficacia del vaccino – Prima insorgenza di COVID-19 a partire dai 7 giorni successivi alla somministrazione della dose 2 per sottogruppo di età – soggetti senza evidenza di infezione pregressa da SARS-CoV-2* entro i 7 giorni successivi alla somministrazione della dose 2 – popolazione valutabile ai fini dell’efficacia (7 giorni) durante il periodo di follow-up controllato verso placebo
Nell’analisi di efficacia aggiornata, l’efficacia del vaccino a mRNA anti-COVID-19 nella prevenzione della prima insorgenza di COVID-19 a partire da 7 giorni dopo la somministrazione della seconda dose, rispetto al placebo, è risultata pari al 91,1% (IC al 95% compreso fra 88,8% e 93,0%) nei partecipanti appartenenti alla popolazione valutabile ai fini dell’efficacia con o senza evidenza di infezione pregressa da SARS-CoV-2. 12 Inoltre, le analisi di efficacia aggiornate per sottogruppi hanno dimostrato stime puntuali di efficacia paragonabili fra sessi, etnie, aree geografiche, e fra partecipanti con comorbilità e obesità, associate a un rischio elevato di COVID-19 severa. Efficacia nei confronti di COVID-19 severa Le analisi di efficacia aggiornate degli endpoint secondari di efficacia supportavano il beneficio del vaccino a mRNA anti-COVID-19 nella prevenzione di COVID–19 severa. A partire dal 13 marzo 2021, l’efficacia del vaccino nei confronti di COVID-19 severa viene presentata unicamente per i partecipanti sia con che senza evidenza di infezione pregressa da SARS-CoV-2 (Tabella 4), poiché il computo dei casi di COVID-19 nei partecipanti senza infezione pregressa da SARS-CoV-2 è risultato pari a quello riscontrato nei partecipanti con o senza infezione pregressa da SARS-CoV-2 sia nel gruppo trattato con vaccino a mRNA anti-COVID-19 che nel gruppo trattato con placebo. Tabella 4. Efficacia del vaccino – Prima insorgenza di COVID-19 severa in soggetti con o senza evidenza di infezione pregressa da SARS-CoV-2 in base alla definizione della Food and Drug Administration (FDA)* dopo la somministrazione della dose 1 oppure a partire da 7 giorni dopo la somministrazione della dose 2 durante il follow-up controllato verso placebo
Efficacia e immunogenicità negli adolescenti di età compresa fra 12 e 15 anni – dopo 2 dosi In un’analisi dello Studio 2 condotta su adolescenti di età compresa fra 12 e 15 anni senza evidenza di infezione pregressa, non sono stati identificati casi tra i 1.005 partecipanti che avevano ricevuto il vaccino, mentre si sono verificati 16 casi tra i 978 partecipanti che avevano ricevuto placebo. La stima puntuale di efficacia risulta pari al 100% (intervallo di confidenza al 95% compreso fra 75,3 e 100,0). Nei partecipanti con o senza evidenza di infezione pregressa si sono verificati 0 casi tra i 1.119 partecipanti che avevano ricevuto il vaccino, e 18 casi tra i 1.110 partecipanti che avevano ricevuto placebo. Anche questo indica che la stima puntuale di efficacia è pari a 100% (intervallo di confidenza al 95% compreso fra 78,1 e 100,0). Nello Studio 2 è stata condotta un’analisi dei titoli degli anticorpi neutralizzanti SARS-CoV-2 1 mese dopo la somministrazione della dose 2 in un sottogruppo di partecipanti, selezionati in modo casuale, che non presentavano evidenza sierologica o virologica di infezione pregressa da SARS-CoV-2 fino a 1 mese dopo la somministrazione della dose 2, per confrontare la risposta negli adolescenti di età compresa fra 12 e 15 anni (n = 190) e quella nei partecipanti di età compresa fra 16 e 25 anni (n = 170). Il rapporto della media geometrica dei titoli anticorpali (GMT) fra la fascia di età compresa fra 12 e 15 anni e la fascia di età compresa fra 16 e 25 anni è risultato pari a 1,76, con un IC al 95% a 2 code compreso fra 1,47 e 2,10. Pertanto, il criterio di non inferiorità fissato a 1,5 volte è risultato soddisfatto, poiché il limite inferiore dell’IC al 95% a due code per il rapporto della media geometrica (GMR) è risultato >0,67. Immunogenicità nei partecipanti di età pari o superiore a 18 anni – dopo la dose di richiamo (terza dose) L’efficacia di una dose di richiamo di Comirnaty si basava sulla misurazione del titolo degli anticorpi neutralizzanti anti-SARS-CoV-2 al 50% (NT50) (USA_WA1/2020).
Nello Studio 2, le analisi dei valori di NT50 1 mese dopo la somministrazione della dose di richiamo rispetto a 1 mese dopo la somministrazione del ciclo primario in soggetti di età compresa fra 18 e 55 anni senza evidenza sierologica o virologica di infezione pregressa da SARS-CoV-2 entro 1 mese dopo la somministrazione della vaccinazione di richiamo hanno dimostrato la non inferiorità sia in termini di rapporto della media geometrica (GMR) che di differenza nei tassi di sierorisposta.
La sierorisposta in un partecipante era definita come il raggiungimento di un aumento ≥4 volte del valore di NT50 rispetto al basale (prima del ciclo primario).
Queste analisi sono riassunte nella Tabella 5.
Tabella 5. Saggio di neutralizzazione di SARS-CoV-2 – NT50 (titolo)† (SARS-CoV-2 USA_WA1/2020) – confronto in termini di GMT e tasso di sierorisposta fra 1 mese dopo la somministrazione della dose di richiamo e 1 mese dopo la somministrazione del ciclo primario – partecipanti di età compresa fra 18 e 55 anni senza evidenza di infezione entro 1 mese dopo la dose di richiamo* – popolazione sottoposta a somministrazione della dose di richiamo, valutabile ai fini dell’immunogenicità
Popolazione pediatrica L’Agenzia europea dei medicinali ha rinviato l’obbligo di presentare i risultati degli studi con Comirnaty nella popolazione pediatrica per la prevenzione di COVID-19(vedere paragrafo 4.2 per informazioni sull’uso pediatrico). Questo medicinale è stato autorizzato con procedura “subordinata a condizioni”.
Ciò significa che devono essere forniti ulteriori dati su questo medicinale.
L’Agenzia europea dei medicinali esaminerà almeno annualmente le nuove informazioni su questo medicinale e il riassunto delle caratteristiche del prodotto (RCP) verrà aggiornato, se necessario. 5.2Proprietà farmacocinetiche Non pertinente. 5.3Dati preclinici di sicurezza I dati preclinici non rivelano rischi particolari per l’uomo sulla base di studi convenzionali di tossicità a dosi ripetute e tossicità della riproduzione e dello sviluppo. Tossicità generale I ratti che avevano ricevuto Comirnaty per via intramuscolare (3 dosi complete destinate all’uomo somministrate una volta a settimana, che generavano livelli relativamente più elevati nei ratti a causa delle differenze di peso corporeo) hanno mostrato edema ed eritema in sede di iniezione, e un incremento dei leucociti (inclusi basofili ed eosinofili) compatibile con una risposta infiammatoria, unitamente a una vacuolizzazione degli epatociti della vena porta, senza evidenza di danno epatico. Tutti gli effetti sono risultati reversibili. Genotossicità/Potenziale cancerogeno Non sono stati condotti studi di genotossicità o sul potenziale cancerogeno. Si ritiene che i componenti del vaccino (lipidi e mRNA) non presentino alcun potenziale genotossico. Tossicità della riproduzione La valutazione della tossicità della riproduzione e dello sviluppo è stata condotta nei ratti nel corso di uno studio combinato di fertilità e tossicità dello sviluppo, durante il quale femmine di ratto hanno ricevuto Comirnaty per via intramuscolare prima dell’accoppiamento e durante la gestazione (4 dosi complete destinate all’uomo, che generavano livelli relativamente più elevati nei ratti a causa delle differenze di peso corporeo, somministrate dal giorno 21 precedente all’accoppiamento fino al giorno 20 della gestazione).
Sono state osservate risposte anticorpali neutralizzanti verso SARS-CoV-2 nelle madri animali da prima dell’accoppiamento al termine dello studio al giorno 21 dopo la nascita, così come nei feti e nella prole.
Non si sono verificati effetti correlati al vaccino in termini di fertilità nelle femmine, gravidanza, sviluppo embrionale/fetale o sviluppo della prole. Non sono disponibili dati su Comirnaty relativi al trasferimento placentare o all’escrezione nel latte materno del vaccino. 6.INFORMAZIONI FARMACEUTICHE 6.1Elenco degli eccipienti ((4-idrossibutil)azanediil)bis(esano-6,1-diil)bis(2-esildecanoato) (ALC-0315) 2-[(polietilenglicole)-2000]-N,N-ditetradecilacetammide (ALC-0159) 1,2-distearoil-sn-glicero-3-fosfocolina (DSPC) Colesterolo Potassio cloruro Potassio diidrogeno fosfato Sodio cloruro Fosfato disodico diidrato Saccarosio Acqua per preparazioni iniettabili Sodio idrossido (per aggiustamento del pH) Acido cloridrico (per aggiustamento del pH) 16 6.2Incompatibilità Questo medicinale non deve essere miscelato con altri medicinali ad eccezione di quelli menzionati nel paragrafo 6.6. 6.3Periodo di validità Flaconcino chiuso Flaconcino congelato 9 mesi se conservato a una temperatura compresa tra -90 °C e -60 °C. Durante il periodo di validità di 9 mesi, i flaconcini chiusi possono essere conservati e trasportati a una temperatura compresa tra -25 °C e -15 °C per un unico periodo di tempo della durata massima di 2 settimane, e possono essere nuovamente riportati a una temperatura compresa tra -90 °C e -60 °C. Flaconcino scongelato 1 mese a una temperatura compresa tra 2 °C e 8 °C durante il periodo di validità di 9 mesi. Durante il periodo di validità di 1 mese a una temperatura compresa tra 2 °C e 8 °C, è consentito il trasporto per un massimo di 12 ore. Prima dell’uso, il flaconcino chiuso può essere conservato fino a 2 ore a una temperatura non superiore a 30 °C. Dopo lo scongelamento, i flaconcini possono essere maneggiati in condizioni di luce ambientale. Una volta scongelato, il vaccino non deve essere ricongelato. Gestione delle escursioni termiche dopo l’estrazione dal congelatore I dati sulla stabilità indicano che il flaconcino chiuso rimane stabile per un massimo di: •24 ore se conservato a temperature comprese tra -3 °C e 2 °C •4 ore in totale se conservato a temperature comprese tra 8 °C e 30 °C; questo intervallo di tempo comprende anche le 2 ore a una temperatura non superiore a 30 °C indicate sopra.
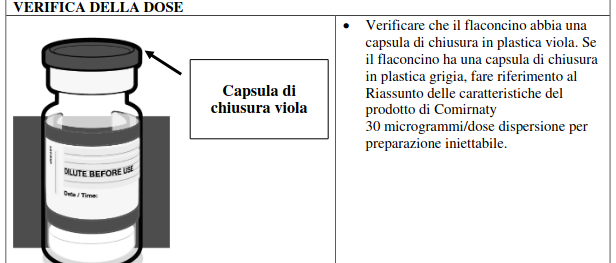
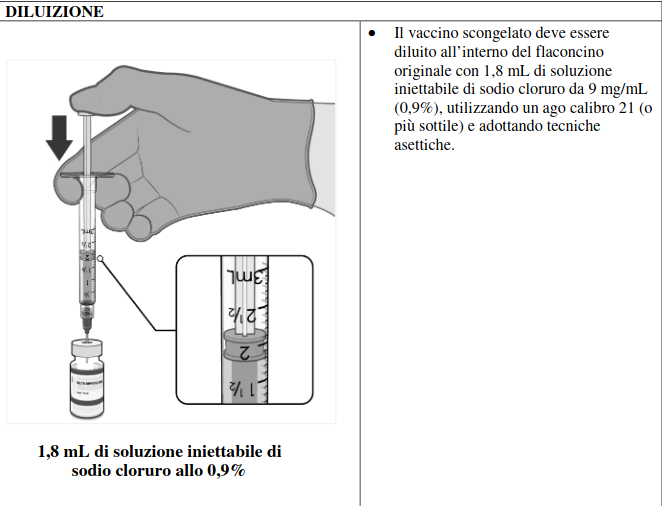
Queste informazioni servono a fornire una guida per gli operatori sanitari solo in caso di escursione termica temporanea. Trasferimenti di flaconcini congelati conservati a temperatura ultra-bassa (<-60 °C) •Una volta estratti dal congelatore a temperatura ultra-bassa (<-60 °C), i vassoi di flaconcini a coperchio chiuso contenenti 195 flaconcini possono rimanere a temperature non superiori a 25 °C per un massimo di 5 minuti. •Una volta estratti dal congelatore a temperatura ultra-bassa (<-60 °C), i vassoi di flaconcini a coperchio aperto o i vassoi di flaconcini contenenti meno di 195 flaconcini possono rimanere a temperature non superiori a 25 °C per un massimo di 3 minuti. •Dopo essere stati nuovamente trasferiti in congelatore in seguito all’esposizione a temperature non superiori a 25 °C, i vassoi di flaconcini devono rimanere in congelatore per almeno 2 ore prima che sia possibile estrarli nuovamente. Trasferimenti di flaconcini congelati conservati a una temperatura compresa tra -25 °C e -15 °C •Una volta estratti dal congelatore (a una temperatura compresa tra -25 °C e -15 °C), i vassoi di flaconcini a coperchio chiuso contenenti 195 flaconcini possono rimanere a temperature non superiori a 25 °C per un massimo di 3 minuti. •Una volta estratti dal congelatore (a una temperatura compresa tra -25 °C e -15 °C), i vassoi di flaconcini a coperchio aperto o i vassoi di flaconcini contenenti meno di 195 flaconcini possono rimanere a temperature non superiori a 25 °C per un massimo di 1 minuto. Quando un flaconcino viene rimosso dal vassoio, deve essere scongelato per l’uso. Medicinale diluito La stabilità chimica e fisica in uso, anche durante il trasporto, è stata dimostrata per 6 ore a una temperatura compresa tra 2 °C e 30 °C in seguito a diluizione con soluzione iniettabile di sodio cloruro da 9 mg/mL (0,9%). Da un punto di vista microbiologico, salvo che il metodo di diluizione escluda il rischio di contaminazione microbica, il prodotto deve essere utilizzato immediatamente. Se non viene utilizzato immediatamente, i tempi e le condizioni di conservazione prima del suo impiego sono di responsabilità dell’operatore. 6.4Precauzioni particolari per la conservazione Conservare in congelatore a una temperatura compresa tra -90 °C e -60 °C. Conservare nella confezione originale per proteggere il medicinale dalla luce. Durante la conservazione, ridurre al minimo l’esposizione alla luce ambientale, ed evitare l’esposizione alla luce solare diretta e alla luce ultravioletta. Per le condizioni di conservazione dopo lo scongelamento e la diluizione vedere paragrafo 6.3. 6.5Natura e contenuto del contenitore 0,45 mL di concentrato in flaconcino multidose trasparente da 2 mL (vetro di tipo I) con tappo (gomma bromobutilica sintetica) e capsula di chiusura rimovibile in plastica viola con sigillo in alluminio. Ogni flaconcino contiene 6 dosi (vedere paragrafo 6.6). Confezione da 195 flaconcini. 6.6Precauzioni particolari per lo smaltimento e la manipolazione Istruzioni per la manipolazione
Comirnaty deve essere preparato da un operatore sanitario adottando tecniche asettiche, per garantire la sterilità della dispersione preparata.
Smaltimento Il medicinale non utilizzato e i rifiuti derivati da tale medicinale devono essere smaltiti in conformità alla normativa locale vigente. 7.TITOLARE DELL’AUTORIZZAZIONE ALL’IMMISSIONE IN COMMERCIO BioNTech Manufacturing GmbH An der Goldgrube 12 55131 Mainz Germania Tel: +49 6131 9084-0 Fax: +49 6131 9084-2121 service@biontech.de 8.NUMERO(I) DELL’AUTORIZZAZIONE ALL’IMMISSIONE IN COMMERCIO EU/1/20/1528/001
9.DATA DELLA PRIMA AUTORIZZAZIONE/RINNOVO DELL’AUTORIZZAZIONE Data della prima autorizzazione: 21 dicembre 2020 10.DATA DI REVISIONE DEL TESTO Informazioni più dettagliate su questo medicinale sono disponibili sul sito web dell’Agenzia europea dei medicinali, http://www.ema.europa.eu







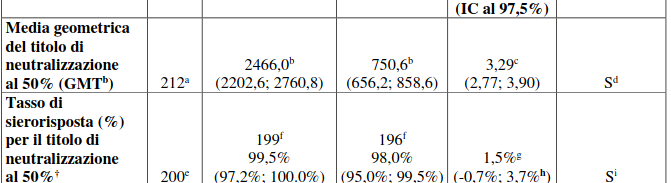






")






![Screenshot 2021-09-25 at 12-04-03 Rêve was 2 days old in the NICU, less than 2 lb and can hold here up straight to look at […]](https://it.nuda-verita.com/wp-content/uploads/2021/09/Screenshot-2021-09-25-at-12-04-03-Reve-was-2-days-old-in-the-NICU-less-than-2-lb-and-can-hold-here-up-straight-to-look-at--150x150.png "Bambini pandemici")


")
